如何用NTsys构建0、1数据(AFLP)进化树

NTSYS-PC使用说明
NTSYS是一个聚类分析的软件,可以用来分析AFLP,RAPD等电泳带型,也可用于微生物群落多样性的相似性分析。下面简单介绍一下其用法:
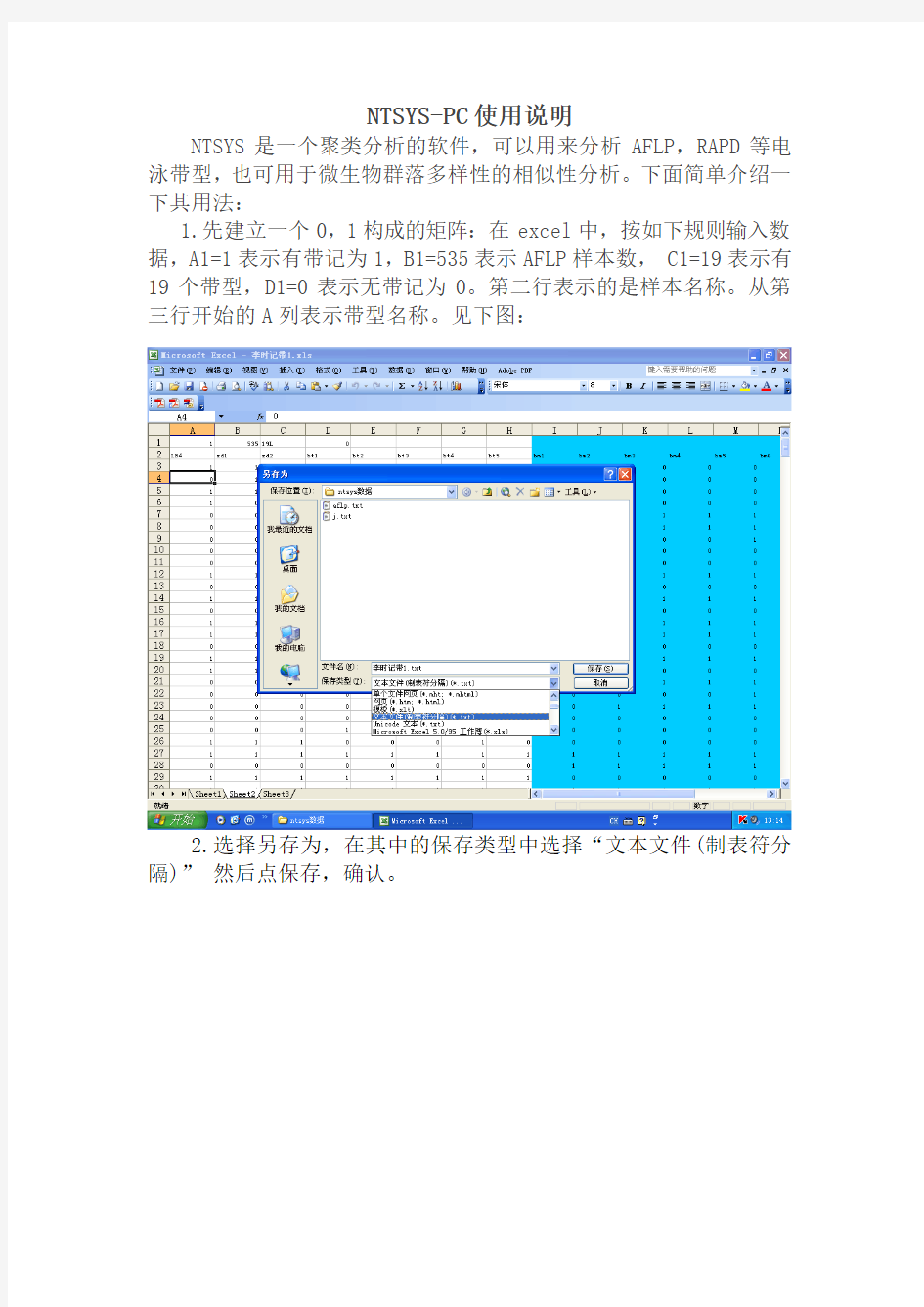
1.先建立一个0,1构成的矩阵:在excel中,按如下规则输入数据,A1=1表示有带记为1,B1=535表示AFLP样本数, C1=19表示有19个带型,D1=0表示无带记为0。第二行表示的是样本名称。从第三行开始的A列表示带型名称。见下图:
2.选择另存为,在其中的保存类型中选择“文本文件(制表符分隔)”然后点保存,确认。
3.打开NTSYS软件点“Similarity”下拉选择”“Qualitative date”在“input file”中选择刚才保存的.txt文件,在“output file”中输入保存文件名。“Byrows”一项不选×,“coefficient”中选择
J,点compute进行运算。
4.点软件左边第二项选择“SAHN”在“input file”中选择上一步运算出来的文件在“output tree file”中输入保存文件名。点compute
进行运算。
5.选择左边第二项中的“Cophenetic Values”在“input tree file”中选择刚才计算的tree文件,输入output的文件名,点compute进
行计算。
6.作Mat检测:点击左面第三项,选择“Matrix comparison plot”在“input file(1)”中选择“Qualitative date”计算出的结果,在“input file(2)”中选择“Cophenetic Valuess”计算出的结果。
点击compute进行计算,r值在0.7以上为可信。
构建进化树方法比较
【转载】分子进化树构建及数据分析的简介 分子进化树构建及数据分析的简介 mediocrebeing, rodger, lylover[1], klaus, oldfish, yzwpf [1] lylover. Email: lylover_2005@https://www.wendangku.net/doc/4918208766.html, 一、引言 开始动笔写这篇短文之前,我问自己,为什么要写这样的文章?写这样的文章有实际的意义吗?我希望能够解决什么样的问题?带着这样的疑惑,我随手在丁香园(DXY)上以关键字“进化分析求助”进行了搜索,居然有289篇相关的帖子(2006年9月12日)。而以关键字“进化分析”和“进化”为关键字搜索,分别找到2,733和7,724篇相关的帖子。考虑到有些帖子的内容与分子进化无关,这里我保守的估计,大约有3,000~4,000篇帖子的内容,是关于分子进化的。粗略地归纳一下,我大致将提出的问题分为下述的几类:1.涉及基本概念。例如,“分子进化与生物进化是不是一个概念”,“关于微卫星进化模型有没有什么新的进展”以及“关于Kruglyak的模型有没有改进的出现”,等等。 2.关于构建进化树的方法的选择。例如,“用boostrap NJ得到XX图,请问该怎样理解?能否应用于文章?用boostrap test中的ME法得到的是XXX树,请问与上个树比,哪个更好”,等等。 3.关于软件的选择。例如,“想做一个进化树,不知道什么软件能更好的使用且可以说明问题,并且有没有说明如何做”,“拿到了16sr RNA数据,打算做一个系统进化树分析,可是原来没有做过这方面的工作啊,都要什么软件”,“请问各位高手用clustalx做出来的进化树与phylip做的有什么区别”,“请问有做过进化树分析的朋友,能不能提供一下,做树的时候参数的设置,以及代表的意思。还有各个分支等数值的意思,说明的问题等”,等等。 4.蛋白家族的分类问题。例如,“搜集所有的关于一个特定domain的序列,共141条,做的进化树不知具体怎么分析”,等等。 5.新基因功能的推断。例如,“根据一个新基因A氨基酸序列构建的系统发生树,这个进化树能否说明这个新基因A和B同源,属于同一基因家族”,等等。 6.计算基因分化的年代。例如,“想在基因组水平比较两个或三个比较接近物种之间的进化年代的远近,具体推算出他们之间的分歧时间”,“如何估计病毒进化中变异所需时间”,等等。 7.进化树的编辑。例如生成的进化树图片,如何进行后续的编辑,比如希望在图片上标注某些特定的内容,等等。 由于相关的帖子太多,作者在这里对无法阅读全部的相关内容而致以歉意。同时,作者归纳的这七个问题也并不完全代表所有的提问。对于问题1所涉及到的基本的概念,作者推荐读者可参考由Masatoshi Nei与Sudhir Kumar所撰写的《分子进化与系统发育》(Molecular Evolution and Phylogenetics)一书,以及相关的分子进化方面的最新文献。对于问题7,作者之一lylover一般使用Powerpoint 进行编辑,而Photoshop、Illustrator及Windows自带的画图工具等都可以使用。
构建系统发育树需要注意的几个问题
构建系统发育树需要注意的几个问题 1 相似与同源的区别:只有当序列是从一个祖先进化分歧而来时,它们才是同源的。 2 序列和片段可能会彼此相似,但是有些相似却不是因为进化关系或者生物学功能相近的缘故,序列组成特异或者含有片段重复也许是最明显的例子;再就是非特异性序列相似。 3 系统发育树法:物种间的相似性和差异性可以被用来推断进化关系。 4 自然界中的分类系统是武断的,也就是说,没有一个标准的差异衡量方法来定义种、属、科或者目。 5 枝长可以用来表示类间的真实进化距离。 6 重要的是理解系统发育分析中的计算能力的限制。任何构树的实验目的基本上就是从许多不正确的树中挑选正确的树。 7 没有一种方法能够保证一颗系统发育树一定代表了真实进化途径。然而,有些方法可以检测系统发育树检测的可靠性。第一,如果用不同方法构建树能得到同样的结果,这可以很好的证明该树是可信的;第二,数据可以被重新取样(bootstrap),来检测他们统计上的重要性。 分子进化研究的基本方法 对于进化研究,主要通过构建系统发育过程有助于通过物种间隐含的种系关系揭示进化动力的实质。 表型的(phenetic)和遗传的(cladistic)数据有着明显差异。Sneath和Sokal(1973)将表型性关系定义为根据物体一组表型性状所获得的相似性,而遗传性关系含有祖先的信息,因而可用于研究进化的途径。这两种关系可用于系统进化树(phylogenetictree)或树状图(dendrogram)来表示。表型分枝图(phenogram)和进化分枝图(cladogram)两个术语已用于表示分别根据表型性的和遗传性的关系所建立的关系树。进化分枝图可以显示事件或类群间的进化时间,而表型分枝图则不需要时间概念。文献中,更多地是使用“系统进化树”一词来表示进化的途径,另外还有系统发育树、物种树(species tree)、基因树等等一些相同或含义略有差异的名称。 系统进化树分有根(rooted)和无根(unrooted)树。有根树反映了树上物种或基
如何做系统进化树
大家好: 我在此介绍几个进化树分析及其相关软件的使用和应用范围。这几个软件分别是PHYLIP、PUZZLE、PAUP、TREEVIEW、CLUSTALX和PHYLO-WIN (LINUX)。 在介绍软件之前,我先简要地叙述一下有关进化树分析的一些方法学问题。进化树也称种系树,英文名叫“Phyligenetic tree”。对于一个完整的进化树分析需要以下几个步骤:⑴要对所分析的多序列目标进行排列(To align sequences)。做ALIGNMENT的软件很多,最经常使用的有CLUSTALX和CLUSTALW,前者是在WINDOW下的而后者是在DOS下的。⑵要构建一个进化树(To reconstrut phyligenetic tree)。构建进化树的算法主要分为两类:独立元素法(discrete character methods)和距离依靠法(distance methods)。所谓独立元素法是指进化树的拓扑形状是由序列上的每个碱基/氨基酸的状态决定的(例如:一个序列上可能包含很多的酶切位点,而每个酶切位点的存在与否是由几个碱基的状态决定的,也就是说一个序列碱基的状态决定着它的酶切位点状态,当多个序列进行进化树分析时,进化树的拓扑形状也就由这些碱基的状态决定了)。而距离依靠法是指进化树的拓扑形状由两两序列的进化距离决定的。进化树枝条的长度代表着进化距离。独立元素法包括最大简约性法(Maximum Parsimony methods)和最大可能性法(Maximum Likelihood methods);距离依靠法包括除权配对法(UPGMAM)和邻位相连法(Neighbor-joining)。⑶对进化树进行评估。主要采用Bootstraping法。进化树的构建是一个统计学问题。我们所构建出来的进化树只是对真实的进化关系的评估或者模拟。如果我们采用了一个适当的方法,那么所构建的进化树就会接近真实的“进化树”。模拟的进化树需要一种数学方法来对其进行评估。不同的算法有不同的适用目标。一般来说,最大简约性法适用于符合以下条件的多序列:i 所要比较的序列的碱基差别小,ii 对于序列上的每一个碱基有近似相等的变异率,iii 没有过多的颠换/转换的倾向,iv 所检验的序列的碱基数目较多(大于几千个碱基);用最大可能性法分析序列则不需以上的诸多条件,但是此种方法计算极其耗时。如果分析的序列较多,有可能要花上几天的时间才能计算完毕。UPGMAM(Unweighted pair group method with arithmetic mean)假设在进化过程中所有核苷酸/氨基酸都有相同的变异率,也就
乳酸菌系统进化树
Lactobacillus.plantarum 204Lactobacillus.pentosus Lactobacillus.paraplantarum 575Lactobacillus.collinoides Lactobacillus.brevis Lactobacillus.farciminis Lactobacillus.alimentarius Lactobacillus.paralimentarius Lactobacillus.kimchii Lactobacillus.sanfranciscensis Lactobacillus.lindneri Lactobacillus.fructivorans Lactobacillus.hilgardii Lactobacillus.parakefiri Lactobacillus.buchneri Lactobacillus.parabuchneri Lactobacillus.kefiri Lactobacillus.kunkeei P.selangorensis Lactobacillus.perolens Lactobacillus.algidus Lactobacillus.mali Lactobacillus.nagelii Lactobacillus.murinus Lactobacillus.animalis Lactobacillus.ruminus Lactobacillus.equi Lactobacillus.agilis Lactobacillus.cypricasei Lactobacillus.acidipiscis Lactobacillus.salivarius Lactobacillus.salicinius Lactobacillus.aviarius Lactobacillus.araffinosus Lactobacillus.coryniformis Lactobacillus.bifermentans Lactobacillus.sakei Lactobacillus.curvatus Lactobacillus.sharpeae Lactobacillus.manihotivorans Lactobacillus.rhamnosus Lactobacillus.zeae Lactobacillus.casei Lactobacillus.panis Lactobacillus.frumenti Lactobacillus.oris Lactobacillus.vaginalis Lactobacillus.pontis Lactobacillus.reuteri Lactobacillus.colehominis Lactobacillus.mucosae Lactobacillus.fermentum Lactobacillus.amylophilus Lactobacillus.johnsonii Lactobacillus.gasseri Lactobacillus.iners Lactobacillus.jensenii Lactobacillus.fornicalis Lactobacillus.psittaci https://www.wendangku.net/doc/4918208766.html,ctis Lactobacillus.delbrueckii Lactobacillus.bulgaricus Lactobacillus.acetotolerans Lactobacillus.hamsteri Lactobacillus.amylolyticus Lactobacillus.intestinalis Lactobacillus.gallinarum Lactobacillus.helveticus Lactobacillus.acidophilus Lactobacillus.crispatus Lactobacillus.amylovorus Lactobacillus.fructosus B.subtilis 99579999 99 704924 98 90 79 999999859996949999 9955 99 85746473999985 999445 404332 67 89 7599 998475999972 6599 5799 52 4798 92 97 91853836481621 59 49 3943 358829 37 12 16 0.01
运用mega5构建系统发生进化树.
1.准备序列文件 准备fasta格式序列文件(fasta格式:大于号>后紧跟序列名,换行后是序列。举例如下)。每条序列可以单独为一个文件,也可以把所有序列放在同一文件内。 核酸序列: >sequence1_name CCTGGCTCAGGATGAACGCT 氨基酸序列: >sequence2_name MQSPINSFKKALAEGRTQIGF 2.多序列比对 打开MEGA 5,点击Align,选择Edit/Build Alignment,选择Create a new alignment,点击OK。
这时需要选择序列类型,核酸(DNA)或氨基酸(Protein)。 选择之后,在弹出的窗口中直接Ctrl + V粘贴序列(如果所有序列在同一个文件中,即可全选序列,复制)。也可以:点击Edit,选择Insert Sequence From File,选择序列文件(可多选)。
序列文件加载之后,呈蓝色背景(为选中状态)。点击按钮,选择Align DNA (如果是氨基酸序列,则会出现Align Protein)。弹出的窗口中设置比对参数,一般都是采用默认参数即可。点击OK,开始多序列比对。
比对完成后,呈现以下状态。 这时需要截齐两端含有---的序列:选中含有---的序列,按键Delete删除(注意:两端都需要截齐)。截齐之后,保存文件为:filename.mas
3.构建系统进化树 多序列比对窗口,点击Data,选择Phylogenetic Analysis,弹出窗口询问:所用序列是否编码蛋白质,根据实际情况选择Yes或No。此时,多序列比对文件就激活了,可以返回MEGA 5主界面建树了。
进化树软件使用综述
建议用Mega相对比较简单些,速度也快点。phylip首先是要用比对好的序列,然后用seeqboot产生bootsrap的序列,然后用prodist(假如是核苷酸序列就用dnadist),然后用neighbor,然后用consense得到最终的树。这个可以参看phylip 的说明文档,还是比较详细的。假如你比对的序列同源度不是太好,可能会有个别步骤报错,好像phylip官方由一个bug修订。假如你熟悉perl的话,我以前写过一个脚本纠正这个bug。。。 在此介绍几个进化树分析及其相关软件的使用和应用范围。这几个软件分别是PHYLIP、PUZZLE、PAUP、TREEVIEW、CLUSTALX和PHYLO-WIN(LINUX)。 在介绍软件之前,我先简要地叙述一下有关进化树分析的一些方法学问题。进化树也称种系树,英文名叫“Phyligenetic tree”。对于一个完整的进化树分析需要以下几个步骤:⑴要对所分析的多序列目标进行排列(To align sequences)。做ALIGNMENT的软件很多,最经常使用的有CLUSTALX和CLUSTALW,前者是在WINDOW下的而后者是在DOS下的。⑵要构建一个进化树(To reconstrut phyligenetic tree)。构建进化树的算法主要分为两类:独立元素法(discrete character methods)和距离依靠法(distance methods)。所谓独立元素法是指进化树的拓扑形状是由序列上的每个碱基/氨基酸的状态决定的(例如:一个序列上可能包含很多的酶切位点,而每个酶切位点的存在与否是由几个碱基的状态决定的,也就是说一个序列碱基的状态决定着它的酶切位点状态,当多个序列进行进化树分析时,进化树的拓扑形状也就由这些碱基的状态决定了)。而距离依靠法是指进化树的拓扑形状由两两序列的进化距离决定的。进化树枝条的长度代表着进化距离。独立元素法包括最大简约性法(Maximum Parsimony methods)和最大可能性法(Maximum Likelihood methods);距离依靠法包括除权配对法(UPGMAM)和邻位相连法(Neighbor-joining)。⑶对进化树进行评估。主要采用Bootstraping法。进化树的构建是一个统计学问题。我们所构建出来的进化树只是对真实的进化关系的评估或者模拟。如果我们采用了一个适当的方法,那么所构建的进化树就会接近真实的“进化树”。模拟的进化树需要一种数学方法来对其进行评估。不同的算法有不同的适用目标。一般来说,最大简约性法适用于符合以下条件的多序列:i 所要比较的序列的碱基差别小,ii 对于序
Mega的使用以及进化树的绘制
1.MEGA构建系统进化树的步骤 2.CLUSTALX进行序列比对 1.MEGA构建系统进化树的步骤 1. 将要用于构建系统进化树的所有序列合并到同一个fasta格式文件,注意:所有序列的方向都要保持一致( 5’-3’)。如图: 2. 打开MEGA软件,选择"Alignment" - "Alignment Explorer/CLUSTAL",在对话框中选择Retrieve sequences from a file, 然后点OK,找到准备好的序列文件并打开,如图: 。 3. 在打开的窗口中选择”Alignment”-“Align by ClustalX” 进行对齐,对齐过程需要一段时间,对齐完成后,最好将序列两端切齐,选择两端不齐的部分,
单击右键,选择delete即可,如图: 。 4. 关闭当前窗口,关闭的时候会提示两次否保存,第一次无所谓,保存不保存都可以,第二次一定要保存,保存的文件格式是.meg。根据提示输入Title,然后会出现一个对话框询问是否是Protein-coding nucleotide sequence data, 根据情况选择Yes或No。最后出现一个对话框询问是否打开,选择Yes,如图: 。 5. 回到MEGA主窗口,在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” -“Neighbor-joining”,打开一个窗口,里面有很多参数可以设
置,如何设置这些参数请参考详细的MEGA说明书,不会设置就暂且使用默认值,不要修改,点击下面的Compute按钮,系统进化树就画出来了,如图: 在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“Minimun-evolution”,如图: 在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“Maximun-parsimony”,如图: 在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“UPGMA”,
分子进化与系统进化树的构建
分子进化与系统进化树的构建 分子进化与系统进化树的构建 分子进化与系统进化树的构建 主要内容: 1、分子进化的研究方法 2、系统进化树的构建方法 3、系统进化树构建常用软件汇集 4、系统进化树构建方法及软件的选择 5、Phylip分子进化分析软件包简介及使用 6、如何利用MEGA3.1构建进化树 声明: 1、本篇涉及的资源主要源于网络及相关书籍,由酷友搜集、分析、整理、审改,供大家学习参考用,如有转载、传播请注明源于基因酷及本篇的工作人员;若本篇侵犯了您的版权或有任何不妥,请Email genecool@https://www.wendangku.net/doc/4918208766.html,告知。 2、由于我们的学识、经验有限,本篇难免会存在一些错误及缺陷,敬请不吝赐教:请到基因酷论坛(https://www.wendangku.net/doc/4918208766.html,/bbs)本篇对应的专题跟贴指出或Email genecool@https://www.wendangku.net/doc/4918208766.html,。 致谢: 整编者:flashhyh 主要参考资料:《生物信息学札记》樊龙江;《分子进化分析与相关软件的应用》作者不详;《进化树构建》ZHAO Yangguo;《如何用MEGA 3.1构建进化树》作者不详;《MEGA3指南》作者不详; 分子进化的研究方法 分子进化的研究方法 分子进化的研究方法 分子进化研究的意义 自20世纪中叶,随着分子生物学的不断发展,进化研究也进入了分子进化(molecularevolution)研究水平,并建立了一套依赖于核酸、蛋白质序列信息的理论和方法。随着基因组测序计划的实施,基因组的巨量信息对若干生物领域重大问题的研究提
供了有力的帮助,分子进化研究再次成为生命科学中最引人注目的领域之一。这些重大问题包括:遗传密码的起源、基因组结构的形成与演化、进化的动力、生物进化等等。分子进化研究目前更多地是集中在分子序列上,但随着越来越多生物基因组的测序完成,从基因组水平上探索进化奥秘,将开创进化研究的新天地。 分子进化研究最根本的目的就是从物种的一些分子特性出发,从而了解物种之间的生物系统发生的关系。通过核酸、蛋白质序列同源性的比较进而了解基因的进化以及生物系统发生的内在规律。 分子进化研究的基础 假设假设::核苷酸和氨基酸序列中含有生物进化历史的全部信息核苷酸和氨基酸序列中含有生物进化历史的全部信息。。 分子钟理论:在各种不同的发育谱系及足够大的进化时间尺度中,许多序列的进化速率几乎是恒定不变的。如下图: 直系同源与旁系同源 直系同源(orthologs):同源的基因是由于共同的祖先基因进化而产生的; 旁系同源(paralogs):同源的基因是由于基因复制产生的。 两者之间的关系如下图所示: 注:用于分子进化分析中的序列必须是直系同源的用于分子进化分析中的序列必须是直系同源的 用于分子进化分析中的序列必须是直系同源的,才能真实反映进化过程。 分子进化研究的基本方法 对于进化研究,主要通过构建系统发育过程有助于通过物种间隐含的种系关系揭示进化动力的实质。 表型的(phenetic)和遗传的(cladistic)数据有着明显差异。Sneath 和Sokal(1973)将表型性关系定义为根据物体一组表型性状所获得的相似性,而遗传性关系含有祖先的信息,因而可用于研究进化的途径。这两种关系可用于系统进化树(phylogenetictree)或树状图(dendrogram)来表示。表型分枝图(phenogram)和进化分枝图(cladogram)两个术语已用于表示分别根据表型性的和遗传性的关系所建立的关系树。进化分枝图可以显示事件或类群间的进化时间,而表型分枝图则不需要时间概念。文献中,更多地是使用“系统进化树”一词来表示进化的途径,另外还有系统发育树、物种树(speciestree)、基因树等等一些相同或含义略有差异的名称. 系统进化树分有根(rooted)和无根(unrooted)树。有根树反映了树上物种或基因的时间顺序,而无根树只反映分类单元之间的距离而不涉及谁是谁的祖先问题。下图表示了
mega5进化树构建
mega5进化树构建 图文详解MEGA 5构建系统发育树 1.准备序列文件 准备fasta 格式序列文件(fasta 格式:大于号>后紧跟序列名,换行后是序列。举例如下)。每条序列可以单独为一个文件,也可以把所有序列放在同一文件内。 核酸序列: >sequence1_name CCTGGCTCAGGATGAACGCT 氨基酸序列: >sequence2_name MQSPINSFKKALAEGRTQIGF 2.多序列比对 打开MEGA 5,点击Align ,选择Edit/Build Alignment,选择Create a new alignment ,点击OK 。 → 这时需要选择序列类型,核酸(DNA )或氨基酸(Protein )。 选择之后,在弹出的窗口中直接Ctrl + V粘贴序列(如果所有序列在同一个文件中,即可全选序列,复制)。也可以:点击Edit ,选择Insert Sequence From File,选择序列文件(可多选)。 序列文件加载之后,呈蓝色背景(为选中状态)。点击按钮,选择Align DNA (如果是氨基酸序列,则会出现Align Protein)。弹出的窗口中设置比对参数,一般都是采用默认参数即可。点击OK ,开始多序列比对。 比对完成后,呈现以下状态。 这时需要截齐两端含有---的序列:选中含有---的序列,按键Delete 删除(注意:两端都需要截齐)。截齐之后,保存文件为:filename.mas ↓ 3.构建系统进化树 多序列比对窗口,点击Data ,选择Phylogenetic Analysis,弹出窗口询问:所用序列是否编码蛋白质,根据实际情况选择Yes 或No 。此时,多序列比对文件就激活了,可以返回MEGA 5主界面建树了。 MEGA 5主界面。点击Phylogeny ,选择Construct/Test
构建系统进化树的方法步骤
构建系统进化树的方法步骤 1. 建树前的准备工作 1.1 相似序列的获得——BLAST BLAST是目前常用的数据库搜索程序,它是Basic Local Alignment Search Tool的缩写,意为“基本局部相似性比对搜索工具”(Altschul et al.,1990[62];1997[63])。国际著名生物信息中心都提供基于Web的BLAST服务器。BLAST算法的基本思路是首先找出检测序列和目标序列之间相似性程度最高的片段,并作为内核向两端延伸,以找出尽可能长的相似序列片段。 首先登录到提供BLAST服务的常用网站,比如国内的CBI、美国的NCBI、欧洲的EBI和日本的DDBJ。这些网站提供的BLAST服务在界面上差不多,但所用的程序有所差异。它们都有一个大的文本框,用于粘贴需要搜索的序列。把序列以FASTA格式(即第一行为说明行,以“>”符号开始,后面是序列的名称、说明等,其中“>”是必需的,名称及说明等可以是任意形式,换行之后是序列)粘贴到那个大的文本框,选择合适的BLAST程序和数据库,就可以开始搜索了。如果是DNA序列,一般选择BLASTN搜索DNA数据库。 这里以NCBI为例。登录NCBI主页-点击BLAST-点击Nucleotide-nucleotide BLAST (blastn)-在Search文本框中粘贴检测序列-点击BLAST!-点击Format-得到result of BLAST。 BLASTN结果如何分析(参数意义): >gi|28171832|gb|AY155203.1| Nocardia sp. ATCC 49872 16S ribosomal RNA gene, complete sequence Score = 2020 bits (1019), Expect = 0.0 Identities = 1382/1497 (92%), Gaps = 8/1497 (0%) Strand = Plus / Plus Query: 1 gacgaacgctggcggcgtgcttaacacatgcaagtcgagcggaaaggccctttcgggggt 60 |||||||||||||||||||||||||||||||||||||||||| ||||||||| ||||| Sbjct: 1 gacgaacgctggcggcgtgcttaacacatgcaagtcgagcggtaaggcccttc--ggggt 58 Query: 61 actcgagcggcgaacgggtgagtaacacgtgggtaacctgccttcagctctgggataagc 120 || ||||||||||||||||||||||||||||||| | |||||| ||||||||||||| Sbjct: 59 acacgagcggcgaacgggtgagtaacacgtgggtgatctgcctcgtactctgggataagc 118 Score :指的是提交的序列和搜索出的序列之间的分值,越高说明越相似;
MEGA构建系统进化树的步骤(以MEGA7为例)
MEGA构建系统进化树的步骤(以MEGA7为例) 本文是看中国慕课山东大学生物信息学课程总结出来的 分子进化的研究对象是核酸和蛋白质序列。研究某个基因的进化,是用它的DNA序列,还是翻译后的蛋白质序列呢?序列的选取要遵循以下原则:1)如果DNA序列的两两间的一致度≥70%,选用DNA 序列。因为,如果DNA序列都如此相似,它的蛋白质会相似到看不出区别,这对构建系统发生树是不利的。所以这种情况下应该选用DNA序列,而不选蛋白质序列。2)如果DNA序列的两两间的一致度≤70%,DNA序列和蛋白质序列都可以选用。 1. 将要用于构建系统进化树的所有序列合并到同一个fasta格式文件,注意:所有序列的方向都要保持一致( 5’-3’)。 想要做系统发生树先要做多序列比对,然后把多序列比对的结果提交给建树软件进行建树,所以在用MEGA建树时可以输入一个已经比对好的多序列比对,也可以输入一条原始序列,让MEGA先来做多序列比对,再建树(一般我们都是原始序列)。所以我们以后者为例。 2.打开MEGA软件,选择主窗口的”File”→“Open A File”→找到并打开fasta文件,这时会询问以何种方式打开,我们是原始序列,需要先进行多序列比对,所以选择“Align”。如果是比对好的多序列比对可以直接选择“Analyze”。 3.在打开的Alignment Explorer窗口中选择”Alignment”-“Align by ClustalW”进行多序列比对(MEGA提供了ClustalW和Muscle两种多序列比对方法,这里选择熟悉的ClustalW),弹出窗口询问“Nothing selected for alignment,Select all?”选择“OK”。 4. 之后,弹出多序列比对参数设置窗口。这个窗口和EMBL在线多序列比对一样,可以设置替换记分矩阵、不同的空位罚分(罚分填写的是正数,计算时按负数计算)等参数。MEGA的所有默认参数都是经过反复考量设置的,这保证了MEGA傻瓜机全自动档的品质,所以当你无从下手,或者没有什么特别要求的时候,直接点击“OK”,接受这些默认参数,开始多序列比对。
系统发育树构建方法优劣
1.邻接法邻接法(neighbor-joiningmethod,NJ)由Saitou和Nei(1987)提出,NJ法是基于最小进化原理经常被使用的一种算法,它不检验所有可能的拓扑结构,能同时给出拓扑结构和分支长度。在重建系统发生树时,它取消了UPGMA法所做的假定,认为在进化分支上,发生趋异的次数可以不同。最近的计算机模拟已表明它是最有效的基于距离数据重建系统树的方法之一。该方法通过确定距离最近(或相邻)的成对分类单位来使系统树的总距离达到最小。它的特点是重建的树相对准确,假设少,计算速度快,只得一棵树。其缺点主要表现在将序列上的所有位点等同对待,且所分析序列的进化距离不能太大。故NJ法适用于进化距离不大,信息位点少的短序列。邻接法在距离建树中经常会用到,而不用理会使用什么样的优化标准。完全解析出的进化树是通过对完全没有解析出的“星型”进化树进行“分解”得到的,分解的步骤是连续不断地在最接近(实际上是最孤立的)的序列对中插入树枝,而保留进化树的终端。于是,最接近的序列对被巩固了,而“星型”进化树被改善了,这个过程将不断重复。这个方法相对而言很快,也就是说,对于一个50个序列的进化树,只需要若干秒甚至更少。 2.最大简约法最大简约法(maximum parsimony method,MP)最早是基于形态特征分类的需要发展起来的,具体的算法有许多不同版本,其中有些已被广泛地应用于分子进化研究中。利用MP方法重建系统发生树,实际上是一个对给定OTUs其所有可能的树进行比较的过程。对某一个可能的树,首先对每个位点祖先序列的核苷酸组成做出推断,然后统计每个位点用来阐明差异的核苷酸最小替换数目。在整个树中,所有信息简约位点最小核苷酸替换数的总和称为树的长度(常青和周开亚,1998)。MP法是一种优化标准,这种标准遵循“奥卡姆剃刀原则(Occam’S Razor principle)”:对数据最好的解释也是最简单的,而最简单的所需要的特别假定也最少。MP法基于进化过程中所需核苷酸(或氨基酸)替代数目最少的假说,对所有可能正确的拓扑结构进行计算并挑选出所需替代数最小的拓扑结构作为最优系统树,也就是通过比较所有可能树,选择其中长度最小的树作为最终的系统发生树,即最大简约树(maximum parsimony tree)。与其他建树方法相比,MP法无需引入处理核苷酸或者氨基酸替代时所必需的假设(替代模型)。同时,MP法对于分析某些特殊的分子数据(如插入序列和插入/缺失)有用。在分析的序列位点上没有回复突变或平行突变,且被检验的序列位点数很大的时候,MP法能够获得正确的(真实)系统树。但MP法推导的树不是唯一的,在分析序列上存在较多的回复突变或平行突变,而被检验的序列位点数又比较少的时候,最大简约法可能会出现建树错误。故MP法适用于序列残基差别小,具有近似变异率,包含信息位点比较多的长序列。 3.最大似然法最大似然法(maximum likelihood method,MI。)是20世纪60年代末期由于对地生物信息学分析实践震波和水声信号等处理的需要而发展起来的一种非线性谱估计方法。最早由凯佩用这种方法对空间阵列接收信号进行频率波数谱估值,后来推广到对时问信号序列的功率谱估值。 最大似然法最早应用于系统发育分析是在对基因频率数据的分析上。其原理是考虑到每个位点出现残基的似然值,将每个位置所有可能出现的残基替换概率进行累加,产生特定位点的似然值。MI。法对所有可能的系统发育树都计算似然函数,似然函数值最大的那棵树即为最可能的系统发育树。利用最大似然法来推断一组序列的系统发生树,需首先确定序列进化的模型,如Jukes—Cantor模型、Kimura二参数模型及一般二参数模型等。在进化模型选择合理的情况下,MI。法是与进化事实吻合最好的建树算法。其缺点是计算强度非常大,极为耗时。
用MEGA构建进化树
如何用MEGA构建进化树 MEGA3、1就是一个关于序列分析以及比较统计得工具包,其中包括有距离建树法与MP建树法;可自动或手动进行序列比对,推断进化树,估算分子进化率,进行进化假设测验,还能联机得Web数据库检索。下载后可直接使用,主要包括几个方面得功能软件:i)DNA与蛋白质序列数据得分析软件。ii)序列数据转变成距离数据后,对距离数据分析得软件。iii)对基因频率与连续得元素分析得软件。iv)把序列得每个碱基/氨基酸独立瞧待(碱基/氨基酸只有0与1得状态)时,对序列进行分析得软件。v)绘制与修改进化树得软件,进行网上blast搜索。 用MEGA构建进化树有以下步骤: 1、16S rDNA测序与参考序列选取 从环境中分离到单克隆,去重复后扩增16S rDNA序列并测序,然后与数据库比对,找到相似度最高得几个序列,确定一下您分离得细菌大约属于哪个科哪个属,如果相似度达到百分之百那基本可以确定您分离得到得就就是Blast到得那个,然后找一到两个同科得,再找一到两个同目得,再找一到两个同纲得细菌,把序列全部下下来,以FSATA形式整合在TXT文档中,如 >TS1 GCAGTCGAACGATGAAGCCCAGCTTGCTGGGTGGA TTAGTGGCGAACGGGTGAGTAACACGTGGGTGATCTGCCCTGCACTTCGGGATAAGCCTGGGAAACTGGGTCTAATACCGGATAGGACCTCGGGA TGCATGTTCCGGGGTGGAAAGGTTTTCCGGTGCAGGATGGGCC >gi|117572706|gb|EF028124、1| Rhodococcus sp、Atl25 16S ribosomal RNAgene,partial sequence CGATTAGAGTTTGATCCTGGCTCAGGACGAACGCTGGCGGCGTGCTTAACACATGCAAGTCGAACGATGAAGCCCAGCTTGCTGGGTGGATTAGTGGCGAACGGGTGAGTAACACGTGGGTGA TCTGCCCTGCACTTCGGGATAAGCCTGGGAAACTGGGTCTAATACCGGAT >TS2 TGCAAGTCGAGCGAATGGA TTAAGAGCTTGCTCTTATGAAGTTAGCGGCGGA CGGGTGAGTAACACGTGGGTAACCTGCCCATAAGACTGGGATAACTCCGG GAAACCGGGGCTAATACCGGATAACATTTTGAACTGCATGGTTCGAAATTGAAAGGCGGCTTCGGCTGTCACT >gi|56383044|emb|AJ809498、1|Bacillus cereus partial16S rRNA gene, strainTMW 2、383 GA TGAACGCTGGCGGCGTGCCTAATACATGCAAGTCGAGCGAATGGATTAAGAGCTTGCTCTTATGAAGTTAGCGGCGGACGGGTGAGTAACACGTGGGTAACCTGCCCATAAGACTGGGA TAACTCCGGGAAACCGGGGCTAA TACCGGATAACA TTTTGAACYGCA TGGTTC…………………………、 …………………………、 参考序列选择有几个原则:a,不选非培养(unclutured)微生物为参比;b,所选参考序列要正确,里面无错误碱基;c,在保证同属得前提下,优先选择16S rDNA全长测序或全基因组测序得种;d,每个种属选择一个参考序列,如果自己得序列中同一属得较多,可适当选择两个参考序列。 2、序列比对
MEGA构建系统进化树的步骤(以MEGA7为例)教学文案
M E G A构建系统进化树的步骤(以M E G A7为 例)
MEGA构建系统进化树的步骤(以MEGA7为例) 本文是看中国慕课山东大学生物信息学课程总结出来的 分子进化的研究对象是核酸和蛋白质序列。研究某个基因的进化,是用它的DNA序列,还是翻译后的蛋白质序列呢?序列的选取要遵循以下原则:1)如果DNA序列的两两间的一致度≥70%,选用DNA序列。因为,如果DNA序列都如此相似,它的蛋白质会相似到看不出区别,这对构建系统发生树是不利的。所以这种情况下应该选用DNA序列,而不选蛋白质序列。2)如果DNA 序列的两两间的一致度≤70%,DNA序列和蛋白质序列都可以选用。 1. 将要用于构建系统进化树的所有序列合并到同一个fasta格式文件,注意:所有序列的方向都要保持一致 ( 5’-3’)。 想要做系统发生树先要做多序列比对,然后把多序列比对的结果提交给建树软件进行建树,所以在用MEGA建树时可以输入一个已经比对好的多序列比对,也可以输入一条原始序列,让MEGA先来做多序列比对,再建树(一般我们都是原始序列)。所以我们以后者为例。 2.打开MEGA软件,选择主窗口的”File”→“Open A File”→找到并打开fasta文件,这时会询问以何种方式打开,我们是原始序列,需要先进行多序列比对,所以选择“Align”。如果是比对好的多序列比对可以直接选择“Analyze”。 3.在打开的Alignment Explorer窗口中选择”Alignment”-“Align by ClustalW”进行多序列比对(MEGA提供了ClustalW和Muscle两种多序列比对方法,这
构建生物进化树的方法比较
极为详细的建树方法,新手入门推荐 生物进化树的构建 目录 前言 (2) 一、 NCBI (6) 二、 Mega (9) 三、 DNAMAN (15) 四、DNAStar (18) 五、 Bio edit (21)
前言 1.背景资料 进化树(evolutionary tree)又名系统树(phylogenetie tree)进化树,用来表示物种间亲缘关系远近的树状结构图。在进化树中,各个分类单元(物种)依据进化关系的远近,被安放在树状图表上的不同位置。所以,进化树简单地表示生物的进化历程和亲缘关系。已发展成为多学科(包括生命科学中 的进化论、遗传学、分类学、分子生物学、生 物化学、生物物理学和生态学,又包括数学中的 概率统计、图论、计算机科学和群论)交叉形成的一个边缘领域。 归纳总结生物进化的总趋势有以下几类: ①结构上:由简单到复杂 ②生活环境上:由水生到陆生 ③进化水平上:由低等到高等 一般来说,进化树是一个二叉树。它由很多的分支和节点构成。根据位置的不同,进化树的节
点分为外部节点和内部节点,外部节点就是我们要进行分类的分类单元(物种)。而物种之间的进化关系则用节点之间的连线表示。内部节点表示进化事件发生的地方,或表示分类单元进化的祖先。在同一个进化树中,分类单元的选择应当标准一致。进化树上不同节点之间的连线称为分支,其中有一端与叶子节点相连的分支称为外枝,不与叶子节点相连的分支称为内枝。 进化树一般有两种:有根树和无根树。有根树有一个鲜明的特征,那就是它有一个唯一的根节点。这个根节点可以理解为所有其他节点的共同祖先。所以,有根树能可以准确地反映各个物种的进化顺序,从根节点进化到任何其他节点只有能有一条惟一的路径。无根树则不能直接给出根节点,无根树只反映各个不同节点之间的进化关系的远近,没有物种如何进化的过程。但是,我们可以在无根树种指派根节点,从而找出各个物种的进化路径。 无根树 有根树
构建系统进化树的详细步骤
构建系统进化树的详细步骤 1. 建树前的准备工作 1.1 相似序列的获得——BLAST BLAST是目前常用的数据库搜索程序,它是Basic Local Alignment Search Tool 的缩写,意 为“基本局部相似性比对搜索工具”(Altschul et al.,1990[62];1997[63])。国际著名生物信息中心 都提供基于Web的BLAST服务器。BLAST算法的基本思路是首先找出检测序列和目标序 列之间相似性程度最高的片段,并作为核向两端延伸,以找出尽可能长的相似序列片段。 首先登录到提供BLAST服务的常用,比如国的CBI、美国的NCBI、欧洲的EBI和日本的DDBJ。这些提供的BLAST服务在界面上差不多,但所用的程序有所差异。它 们都有一个大的文本框,用于粘贴需要搜索的序列。把序列以FASTA格式(即第一行为说明 行,以“>”符号开始,后面是序列的名称、说明等,其中“>”是必需的,名称及说明等可以是 任意形式,换行之后是序列)粘贴到那个大的文本框,选择合适的BLAST程序和数据库,就 可以开始搜索了。如果是DNA序列,一般选择BLASTN搜索DNA数据库。 这里以NCBI为例。登录NCBI主页-点击BLAST-点击Nucleotide-nucleotide BLAST (blastn)-在Search文本框中粘贴检测序列-点击BLAST!-点击Format-得到result of BLAST。 BLASTN结果如何分析(参数意义): >gi|28171832|gb|AY155203.1| Nocardia sp. ATCC 49872 16S ribosomal RNA gene, complete sequence Score = 2020 bits (1019), Expect = 0.0 Identities = 1382/1497 (92%), Gaps = 8/1497 (0%) Strand = Plus / Plus