MOE分子对接教程

MOE分子对接教程
1.在MOE窗口open打开一个有小配体的蛋白(PDB或moe文件)
点击Compute→prepare→Protonate3D(使质子化)
点击OK。打开SEQ面板
删去水分子和与小配体无关的其他杂配体或离子
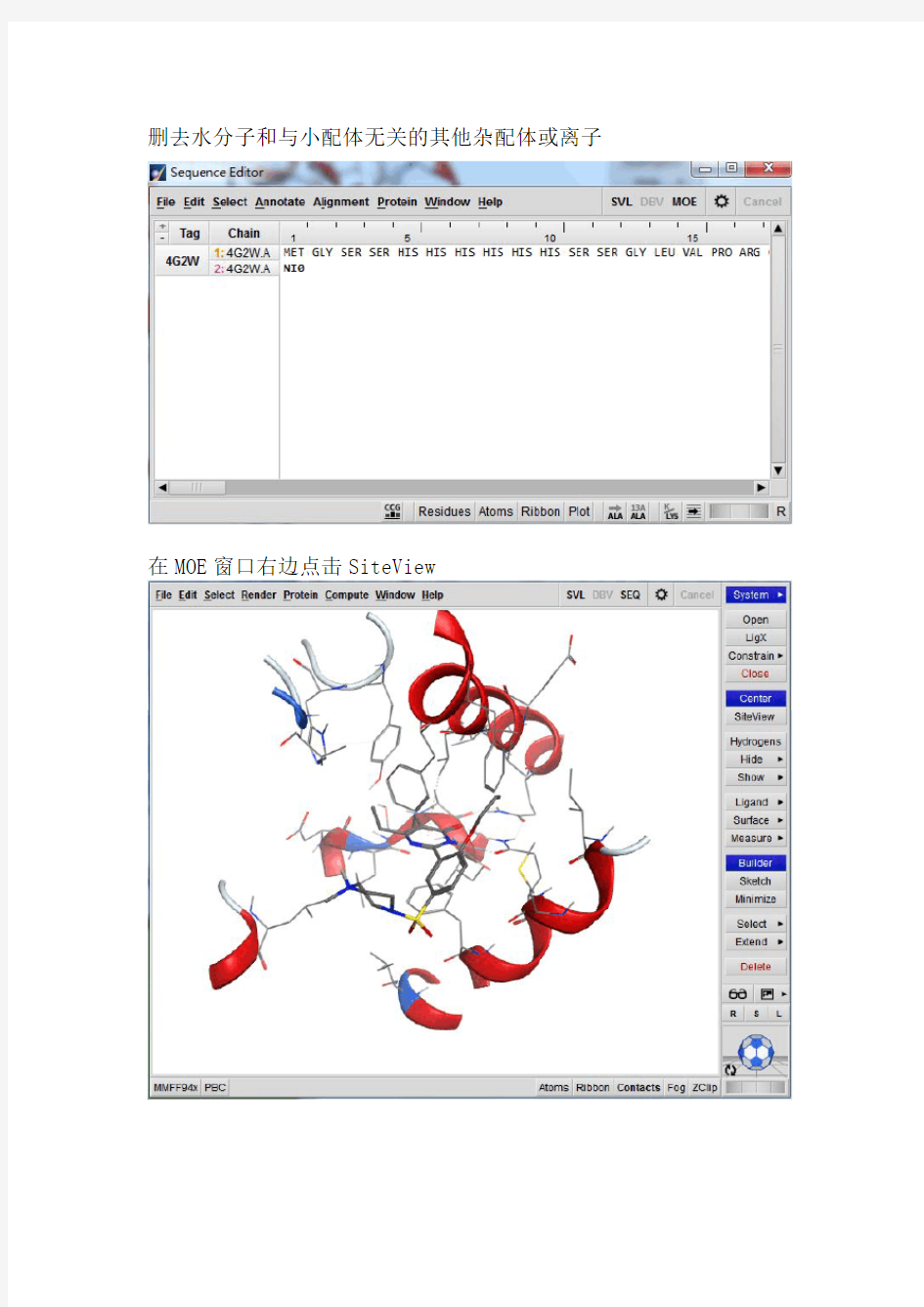
在MOE窗口右边点击SiteView
点击System,改变配体颜色
使得小配体显示绿色C骨架
在MOE窗口点击Surface→recepter
在MOE窗口点击Surface→Surface and Maps 改变口袋表面的透明度
在Surface and Maps面板调节如图所示
在MOE窗口会看到如下图所示:
接下来对接一个小配体
点击Compute→dock
在Recepter选项选择Recepter+Solvent 其它选择默认值。
点击Run。
在表单你会看到有不同的构象对接结果,包括RMSD值。
口袋对接结果
2.创建一个药效团模型
在MOE窗口点击Compute→Pharmacophore→Query Editor
点击R,这样会创建基于受体的药效团特征。
点击口袋中显示的小球,然后点击feature,这样在对接口袋就会显示药效团。
3.化合物数据库的对接
关闭Pharmacophore Query Editor面板
在MOE窗口点击Compute→Dock
Output选项选择一个对接结果数据库名字,这里命名为moe_dock2 Receptor选项为Recepter+Solvent
Ligand选项为MDB File,选择要对接的pde5inhibitors.mdb
(这里说明一下,mdb格式是MOE独有的,用的不多,但是可以将常用的sdf和mol2格式转化为mdb格式,方法为将sdf或mol2文件用MOE打开成表单,再save as成mdb格式。)
其他值默认,点击Run。等待计算...
对接完后,关闭药效团编辑器面板
分子对接
AutoDock和AutoDock Tools 使用教程 一、分子对接简介及软件介绍 1.分子对接理论基础 所谓分子对接就是两个或多个分子之间通过几何匹配和能量匹配而相互识别的过程。分子对接在酶学研究以及药物设计中具有十分重要的意义。在酶激活剂、酶抑制剂与酶相互作用以及药物分子产生药理反应的过程中,小分子(通常意义上的Ligand)与靶酶(通常意义上的Receptor)相互互结合,首先就需要两个分子充分接近,采取合适的取向,使两者在必要的部位相互契合,发生相互作用,继而通过适当的构象调整,得到一个稳定的复合物构象。通过分子对接确定复合物中两个分子正确的相对位置和取向,研究两个分子的构象,特别是底物构象在形成复合物过程中的变化,是确定酶激活剂、抑制剂作用机制以及药物作用机制,设计新药的基础。 分子对接计算是把配体分子放在受体活性位点的位置,然后按照几何互补、能量互补化学环境互补的原则来实时评价配体与受体相互作用的好坏,并找到两个分子之间最佳的结合模式。分子对接最初思想起源于Fisher E.的“锁和钥匙模型”(图),认为“锁”和“钥匙”的相识别的首要条件是他们在空间形状上要互相匹配。然而,配体和受体分子之间的识别要比“锁和钥匙”模型复杂的多。首先,配体和受体分子的构象是变化的,而不是刚性的,配体和受体在对接过程中互相适应对方,从而达到更完美的匹配。其次,分子对接不但要满足空间形状的匹配,还要满足能量的匹配。配体和受体之间的通过底物分子与靶酶分子能否结合以及结合的强度最终是由形成此复合物过程的结合自由能变化ΔG bind所决定的。 互补性(complementarity)和预组坦织(pre-organization)是决定分子对接过程的两个重要原则,前者决定识别过程的选择性,而后者决定识别过理的结合能力。互补性包括空间结构的互补性和电学性质的互补性。1958年Koshland提出了分子识别过程中的诱导契合(induced fit)概念,指出配体与受体相互结合时,受体将采取一个能同底物达到最佳结合的构象(图1)。而受体与配体分子在识别之前将受体中容纳配体的环境组织的越好,其溶剂化能力越低,则它们的识别效果越佳,形成的复合物也就越稳定。
AutoDock分子对接_中文
分子对接 ——使用AutoDock和AutoDock Tools 一、分子对接简介及软件介绍 二、对接准备及对接操作 三、结果分析 一、分子对接简介及软件介绍 1.分子对接理论基础 所谓分子对接就是两个或多个分子之间通过几何匹配和能量匹配而相互识别的过程。分子对接在酶学研究以及药物设计中具有十分重要的意义。在酶激活剂、酶抑制剂与酶相互作用以及药物分子产生药理反应的过程中,小分子(通常意义上的Ligand)与靶酶(通常意义上的Receptor)相互 分子正确的相对位置和取向,研究两个分子的构象,特别是底物构象在形成复合物过程中的变化,是确定酶激活剂、抑制剂作用机制以及药物作用机制,设计新药的基础。 互补的原则来实时评价配体与受体相互作用的好坏,并找到两个分子之间最佳的结合模式。分子对接最初思想起源于Fisher E.的“锁和钥匙模型”(图),认为“锁”和“钥匙”的相识别的首要条件是他们在空间形状上要互相匹配。然而,配体和受体分子之间的识别要比“锁和钥匙”模型复杂的多。首先,配体和受体分子的构象是变化的,而不是刚性的,配体和受体在对接过程中互相适应 程的结合自由能变化ΔG bind所决定的。 互补性(complementarity)和预组坦织(pre-organization)是决定分子对接过程的两个重要原则,前者决定识别过程的选择性,而后者决定识别过理的结合能力。互补性包括空间结构的互补性和电学性质的互补性。1958年Koshland提出了分子识别过程中的诱导契合(induced fit)概念,指出配体与受体相互结合时,受体将采取一个能同底物达到最佳结合的构象(图1)。而受体与配体分子在识别之前将受体中容纳配体的环境组织的越好,其溶剂化能力越低,则它们的识别效果越佳,形成的复合物也就越稳定。
(完整版)10分钟教你掌握分子对接模拟软件(医药向)
首先介绍一下自己吧,本人毕业于南方某知名211大学药学系,目前于澳门科技大学攻读硕士研究生。从本科开始自己就在接触CADD(计算机辅助药物设计)方面的软件知识,在此将分享一些自己的纯干货!下面将以一个实例操作带大家迅速认识和掌握分子模拟对接,希望给各位从事医药行业和药物化学合成的同学带来帮助。 话不多说,下面进入正题。 首先我们搞清楚一个概念:什么是分子模拟对接。分子模拟对接简单来说就是利用电脑软件将受体蛋白与配体分子进行模拟对接,计算它们的结合能(KJ/MOL)大小来判断结合是否紧密,若结合效果比较理想,那么该蛋白受体或配体则是我们理想的分子,可以进一步进行实验室操作,避免盲目实验带来的人力经济损失。 接下来我将介绍一下本篇文章的主角,也是我们所要用到的软件PyRx、Chemdraw、AutodockTools以及PyMol。为了便于理解,简要概括之:Chemdraw为化合物分子绘图软件;PyRx为Autodock Vina算法搭载软件,能够调用其算法直接进行模拟对接;AutodockTools是PyMol为对接结果成像软件,可以进一步分析其结构。 下面正式进入正题,我将大致分为三个板块来进行推进:受体配体的准备;分子对接;结果分析。研究类型为:已知若干配体分子结构,通过受体蛋白测试配体分子活性。 本次筛选意在以COMT酶为受体,从20种与常见氨基酸形成环二肽的目标化合物中筛选出与COMT酶受体结合最为紧密的一种环二肽结构,大大减少了随机筛选的盲目性,有利于进一步研究该类化合物分子的生物学活性与改造成抗帕金森疾病前药的可能。图1展示了20种不同环二肽结构物质的统一结构,随着R基团的不同,所对应的氨基酸也不同。而表1则展示了20种不同环二肽的分子式。 图1 Cycol[DOPA(6-NO2)-AA]
分子对接的原理,方法及应用
分子对接的原理,方法及应用 (PPT里弄一些分子对接的照片,照片素材文件里有) 分子对接 是将已知三维结构数据库中的分子逐一放在靶标分子的活性位点处。通过不断优化受体化合物的位置、构象、分子内部可旋转键的二面角和受体的氨基酸残基侧链和骨架,寻找受体小分子化合物与靶标大分子作用的最佳构象,并预测其结合模式、亲和力和通过打分函数挑选出接近天然构象的与受体亲和力最佳的配体的一种理论模拟分子间作用的方法。 通过研究配体小分子和受体生物大分子的相互作用,预测其亲和力,实现基于结构的药物设计的一种重要方法。 原理: 按照受体与配体的形状互补,性质互补原则,对于相关的受体按其三维结构在小分子数据库直接搜索可能的配体,并将它放置在受体的活性位点处,寻找其合理的放置取向和构象,使得配体与受体形状互补,性质互补为最佳匹配 (配体与受体结合时,彼此存在静电相互作用,氢键相互作用,范德华相互作用和疏水相互作用,配体与受体结合必须满足互相匹配原则,即配体与受体几何形状互补匹配,静电相互作用互补匹配,氢键相互作用互补匹配,疏水相互作用互补匹配) 目的: 找到底物分子和受体分子的最佳结合位置 问题: 如何找到最佳的结合位置以及如何评价对接分子之间的结合强度 方法: 1、首先建立大量化合物的三维结构数据库 2、将库中的分子逐一与靶分子进行“对接” 3、通过不断优化小分子化合物的位置以及分子内部柔性键的二面角,寻找小分子化合物与靶标大分子作用的最佳构象,计算其相互作用及结合能 4、在库中所有分子均完成了对接计算之后,即可从中找出与靶标分子结合的最佳分子 应用: 1)直接揭示药物分子和靶点之间的相互作用方式 2)预测小分子与靶点蛋白结合时的构象 3)基于分子对接方法对化合物数据库进行虚拟筛选,用于先导化合物的发现
分子对接简要介绍
分子对接简介 分子对接(molecular docking)是通过研究小分子配体与受体生物大分子相互作用,预测其结合模式和亲和力进而实现基于结构的药物设计的一种重要的方法。其本质是两个或多个分子之间的识别过程,其过程涉及分子之间的空间匹配和能量匹配。 分子对接的基本原理 分子对接的最初思想起源于Fisher E提出的“锁和钥匙模型”,即受体与配体的相互识别首要条件是空间结构的匹配。 分子对接锁和钥匙模型 分子对接方法的两大课题是分子之间的空间识别和能量识别。空间匹配是分子间发生相互作用的基础,能量匹配是分子间保持稳定结合的基础。对于空间匹配的计算,通常采用格点计算、片断生长等方法,能量计算则使用模拟退火、遗传算法等方法。各种分子对接方法对体系均有一定的简化,根据简化的程度和方式,可以将分子对接方法分为三类: 刚性对接:刚性对接方法在计算过程中,参与对接的分子构像不发生变化,仅改变分子的空间位置与姿态,刚性对接方法的简化程度最高,计算量相对较小,适合于处理大分子之间的对接。比较有代表性的是Wodak和Janin研发的分子对接算法和Jiang等发展的软对接(soft dock)方法。 半柔性对接:半柔性对接方法允许对接过程中小分子构像发生一定程度的变化,但通常会固定大分子的构像,另外小分子构像的调整也可能受到一定程度的限制,如固定某些非关键部位的键长、键角等,半柔性对接方法兼顾计算量与模型的预测能力,是应用比较广泛的对接方法之一。由于小分子相对较小,因此在一定程度考察柔性的基础上,仍可以保持很高的计算效率,在药物设计中,特别是在基于分子对接的数据库搜索中,多采用半柔性分子方
分子对接步骤(详细)
软件安装: 将D软件中pymol-1.5.0.3.win32-py2.7文件夹中的文件按序号安装,安装3_mgltools_win32_1.5.6_Setup.exe文件时如出错,则直接点开U盘中的mgltools_win32_1.5.6_Setup.exe安装,一直安装到5. 安装后将pymol27 解压后复制到C盘将原有的pymol27文件替换 新建一个文件夹存储要做分子对接的“E盘科研,实验方法,分子对接饶燊强,P2X4 and 青藤碱”这个不行,因为文件名要在英文输入状态,不能有空格,不能有中文,而且要区分大小写E/dock/P2X4_s inomenine 在C盘打开Program files (x86),打开The Scripps Research Institute文件夹,打开Autodock,打开4.2.6 将autogrid4.exe,autodock4.exe和The Scripps Research Institute文件夹中Vina中的vina.exe这三个文件同时复制到新建的存储文件夹中“E盘科研,实验方法,分子对接饶燊强,P2X4 and 青藤碱” 1 用pymol打开E/dock/P2X4_sinomenine中的P2X4.pdb文件 Display sequence 找到ATP和其他天然配体分子(绿色的)
去水加氢后直接将4DW1.pdb命名为P2X4.pdb 保存为P2X4.pdb 2打开软件autodock tools ,打开受体文件处理过的文件P2X4.pdb 受体
计算吉布斯能:菜单栏Edit ——Charges ——Compute Gasteiger 转换文件格式:ADT4.2 菜单Grid——Macromolecule——Choose——P2X4 选择p2x4 后,弹出一个提醒框 点击确认,跳出一个文件保存框,把文件保存到工作文件夹(注意:在这里在命名后加上”.pdbqt”)就是P2X4.pdbqt 配体
药物分子与生物相关物质之间的相互作用研究具有非常重要的意义
药物分子与生物相关物质相互作用的方法学研究及其在药 物分析中的应用 学院:生命科学学院 班级:2014级生科(1)班 姓名:胡瑞瑞 学号:2014506066
药物分子与生物相关物质相互作用的方法学研究及其在药物分 析中的应用 药物分子与生物相关物质之间的相互作用研究具有非常重要的意义。随着研究者研究水平的不断提高,分析仪器的不断更新和新药的不断问世,药物与生物分子之间的研究方法也在不断的增多和更新。本论文主要包括分子间相互作用研究的重要意义,分子间相互作用的体系分类,分子间相互作用研究的方法及特点,药物分子与生物相关物质间相互作用。 分子间相互作用的研究意义: 分子一旦形成后就处于其间相互作用的力场之中,而这种力场在很大程度上会影响物质的性质和功能。在生物学中,分子间相互作用[1]是形成高度专一性识别、反应、调控、运输等过程的基础。诸如底物与受体蛋白的结合识别、酶反应,分子信息的读出、免疫学的抗体一抗原结合、DNA结合蛋白的基因表达的调控、基因编码的翻译和转录、病毒进入细胞及细胞识别等。当然这些过程在化学体系、环境体系中也广泛存在,涉及金属离子一配体、酸一碱、细菌一药物、污染物等诸多方面,只要研究的内容涉及两个或多个化学物种通过分子或局部间的弱相互作用力选择性结合或位点识别,均可看成此领域研究的范畴。故主客体相互作用研究的领域已经渗透到包括生命科学、环境科学、分子生物学、配位化学、超分子化学等前沿领域。 相互作用的一方通常被称为受体(主体),另一方被称为配体(客体)。相互作用研究则是获得受体与配体之间相互作用前后生物学的、化学的、物理化学的、分子生物学等性质的变化信息,从而对受体和配体之间的相互作用进行表征与测量。受体与配体之间相互作用的表达方式有多种,包括结合的配比、结合常数、结合位点、作用方式、自由能变等参数,其中表征相互作用强弱最重要的参数之一要算结合常数。从广义上说,主客体的相互作用相当于各层次的物质间各种力的相互关系,包括小分子之间、大分子之间、小分子与大分子及分子组装体之间的结合。其相互作用方式包括共价作用和非共价作用,其中非共价键力的弱相互作用力包括范德华力、亲水一疏水相互作用、静电力和氢键等。 在药学和细胞生物学等研究领域中,测量分子间相互作用的强弱也是一个重要的研究内容,由此可对药物药效进行预测和对污染物毒性进行评价。因此,分
autodock分子对接操作步骤(自总结)
写在文前:经过几周的努力,我终于安装成功并顺利运行了autodock,感谢网上大神的分享,本人根据自己的经验,写了一篇流程,供大家参考。 第一步、准备小分子和大分子的PDB文件 小分子配体的准备:方法有挺多种的,我自己用的方法是先用chemdraw画出小分子结构,然后存为mol格式,然后用chem3D打开这个mol格式的文件,另存为pdb文件就可以了。 大分子受体的准备:一般就是在rcsb这个网站上进行下载,一般蛋白都有不同的来源,比如植物来源或者微生物来源,根据自己的需要进行下载就好,这个有现成的PDB格式,不需要另外的转化。 另外一般从网站上下载的大分子受体中含有溶剂和原配体小分子,需要进行删除,我使用的方法是用pymol打开蛋白受体,执行两个命令remove solvent(去除溶剂)和remove organic(去除小分子)即可。 下述步骤如果没有特别说明,弹出的对话框都是直接点击yes/ok/确定/accept 第二步、准备小分子和大分子的PDBQT文件 大分子的PDBQT文件准备:需要五个步骤 1、打开大分子:file-read molecule,打开准备好的蛋白pdb文件 2、加氢:就是给受体蛋白加上氢原子,edit-hydrogens-add 3、计算电荷:edit-charges-compute gaseigter,这一步就是把所有电荷都调整为整数 4、设定原子类型:edit-atoms-assign AD4 type 5、输出pdbqt文件:file-save-write pdbqt,记得命名的时候手动加上pdbqt后缀名,下面的gpf、dpf这些文件也是一样的。
分子对接软件AutoDockVina在太湖之光操作系统上的移植-最新资料
分子对接软件AutoDockVina在太湖之光操作系统上的移植 1 “神威?太湖之光”计算系统 “神威?太湖之光”计算系统是国家“863计划”重大专项研究成果,是我国第一台全部采用国产处理器构建的超级计算机,由国家并行计算机工程技术研究中心研制[1]。在2016年6月20日世界TOP500超级计算机排名中,“神威?太湖之光”系统峰值运算性能(125.436PFlops)、持续运算性能 (93.015PFlops)、性能功耗比(6.05GFlops/W)三项关键指标均位居世界第一。 “神威?太湖之光”计算系统共包含了40 960个“申威26010”众核处理器。“申威26010”是由国家“核高基”重大专项支持的我国第一款自主研发的众核处理器,由国家高性能集成电路设计中心研制,性能国际领先,并成功量产,打破了美国对我国的技术封锁。处理器基于申威(SW-64)指令集,采用片上融合异构众核架构和FCBGA3832封装,单个处理器包含了260个运算核心。“神威?太湖之光”具有世界领先水平的超大规模系统低功耗控制技术和高密度组装,比目前世界排名第二的系统节能60%以上,单机仓组装密度居世界第一。 同时,基于“神威?太湖之光”系统自主研发软件,建立了基于申威CPU的高性能计算软件生态链。目前“神威?太湖之
光”计算系统开始应用于四个关键领域:先进制造业应用(CFD、CAE)、地球系统建模和天气预报、生物医药领域的计算、大数据分析。 2 分子对接软件AutoDockVina AutoDock是一款开源的分子模拟软件,最主要应用于执行配体―蛋白分子对接[2~3]。它由Scripps研究所的Olson实验室开发与维护,目前最新版本为AutoDock 4.2。AutoDockVina 也是一款由MGL实验室开发的分子对接软件。与AutoDock 4.0相比,AutoDockVina提高了结合模式预测的平均准确度,通过使用更简单的打分函数加快了搜索速度,并且在处理约20个可旋转键的体系时仍然能提供重现性较好的对接结果。 3 AutoDockVina在太湖之光上的编译 “神威?太湖之光”计算系统采用基于Linux的神威Raise OS 2.0.5作为操作系统,多核心处理器的基本软件堆栈包括基本的编译器组件,包括C、C++和Fortran编译器。C语言,支持C99标准。C++语言,支持C++03标准,并提供支持C++11标准的SWGCC编译环境(从核不支持C++)。Fortran语言,支持Fortran2003标准中主要的功能,满足实际课题需求。 第一步,下载AutoDockVina源码[4]。从Scripps研究所的官网下载AutoDockVina源码,最新的版本是1_1_2。 第二步,安装Boost库[5]。Boost库是一个可移植、提供源代码的C++库,作为标准库的后备,是C++标准化进程的开发
分子对接
网址:https://www.wendangku.net/doc/bd10780099.html,/s/blog_602a741d0100lw4g.html 分子对接 分类:AUTODOCK 标签: 杂谈 一:概述 分子对接是指两个或多个分子通过几何匹配和能量匹配相互识别的过程,在药物设计中有十分重要的意义。药物分子在产生药效的过程中,需要与靶酶相互结合,这就要求两个分子要充分接近并采取合适的取向以使二者在必要的部位相互契合,发生相互作用,继而通过适当的构象调整,得到一个稳定的复合物构象。通过分子对接确定复合物中两个分子正确的相对位置和取向,研究两个分子的构象特别是底物构象在形成复合物过程的变化是确定药物作用机制,设计新药的基础。 分子对接计算把配体分子放在受体活性位点的位置,然后按照几何互补、能量互补以及化学环境互补的原则来评价药物和受体相互作用的好坏,并找出两个分子之间最佳的结合模式。由于分子对接考虑了受体结构的信息以及受体和药物分子之间的相互作用信息,因此从原理上讲,它比仅仅从配体结构出发的药物设计方法更加合理。同时,分子对接筛选的化合物库往往采用的是商用数据库,比如可用化合物数据库(ACD)、剑桥晶体结构数据库(CSD)、世界药物索引(WDL)、药用化合物数据库(CMC)以及可用化合物搜索数据库(ACDSC)等等,因此筛选出来的化合物都为已知化合物,而且相当大一部份可以通过购买得到,这为科研提供了很大的方便,近年来,随着计算机技术的发展、靶酶晶体结构的快速增长以及商用小分子数据库的不断更新,分子对接在药物设计中取得了巨大成功,已经成为基于结构药物分子设计中最为重要的方法。 分子对接的最初思想源自于“锁和钥匙”的模型,即“一把钥匙开一把锁”。不过分子对接, 也就是药物分子和靶酶分子间的识别要比“钥匙和锁”的模型要复杂的多,首先表现在药物分子和靶酶分子是柔性的,这样就要求在对接过程中要相互适应以达到最佳匹配;再者,分 子对接不仅要满足空间形状的匹配,还要满足能量的匹配,底物分子与靶酶分子能否结合 以及结合的强度最终是由形成此复合物过程的结合自由能的变化值决定。互补性和预组织是
实验七、基于AutoDock的分子对接实验_wyp
实验七、基于AutoDock的分子对接实验 一、实验目的: 通过本实验了解分子对接的基本原理及分子对接技术在药物分子设计中的应用,学会用AutoDock软件计算大分子受体与小分子配体的结合模式。 二、实验原理: 分子对接是通过研究小分子配体与生物大分子受体的相互作用,预测其结合模式和亲和力进而实现基于结构的药物分子设计(SBDD)的一种重要方法。根据配体与受体作用的“锁匙原理”,分子对接可以有效地确定与靶受体活性部位空间和电性特征互补匹配地小分子化合物。目前,分子对接技术已广泛应用于SBDD中数据库搜寻及虚拟组合库地设计和筛选研究中。此外,分子对接方法还进一步为探讨蛋白质与药物分子间地相互作用提供有效地研究手段。 根据对接过程中是否考虑研究体系的构象变化,可将分子对接方法分为以下三类: ①刚性对接:指在对接过程中,研究体系的构象不发生变化。 ②半柔性对接:指在对接过程中,研究体系尤其是配体的构象允许在一定的范围内变化。适合处理大分子和小分子间的对接,对接过程中,小分子的构象一般是可以变化的,但大分子是刚性的。 ③柔性对接:指在对接过程中,研究体系的构象基本上可以自由变化的。一般用于精确考虑分子间的识别情况。由于计算过程中体系的构象可以变化,所以计算耗费最大。 目前应用较为广泛的分子对软件包括:Dock, AutoDock, FlexX, Gold, Glide, ICM, Surflex, LigandFit等。其中,AutoDock因其预测精度高,对学术用户免费而被广泛应用。本实验将采用AutoDock进行分子对接实验。 AutoDock 是由Scripps Research Institute 开发的一款用于预测生物大分子与其配体之间相互作用的分子对接软件,主要用于小分子与其受体的对接,但也可用于小肽和其受体的对接。Autodock 采用模拟退火和遗传算法来寻找受体和配体最佳的结合位置,用半经验的自由能计算方法来评价受体和配体之间的匹配情况。最新的版本为4.2。
DOCK进行分子对接的步骤
用DOCK做分子对接计算的基本步骤 一、受体文件和配体文件的准备 需要用到Chimera软件(下载网址https://www.wendangku.net/doc/bd10780099.html,/chimera)。1.受体的准备: 用Chimera打开受体文件,没去除配体和水的要先去除配体和水(在Select 选单中选择相应结构直接删掉即可)。然后,选择Tools ->Structure Editi ng ->Dock Prep,对受体分子进行处理,如下图。 如果受体文件参数不全,则不能使用Dock Prep模块,这时需手动选择Struc ture Editing中的AddH和Add Charge对分子进行处理,并保存为mol2格式文件。 之后,删除受体文件中所有的氢,并将重原子保存为pdb文件。 2.配体的准备: 手动给配体加H,加电子,并将配体保存为mol2文件。 二、生成负模 1.生成靶蛋白的分子表面: 需要用到dms程序,命令:“dms rec_noH.pdb -n -w 1.4 -v -o rec.ms”,参数如下: -a #使用所有原子,而非仅仅是氨基酸的原子 -d #改变点的密度 -g #输出到文件 -i #只计算指定原子所构成的表面 -n #计算表面点的垂直面 -w #改变探针半径 -v #详细输出 -o #指定输出文件名称 (必须的) 2. 生成负模 可以使用DOCK中附带的sphgen程序去生成对接需要的球形负模(需要一个I
NSPH文件,DOCK中的Demo里有吧,复制过来就OK),输入命令sphgen就可以了,得到rec.sph。 3.选择一簇负模进行对接计算 运行命令“showsphere < sphgen_cluster.in”将sph文件转换成pdb文件。其输入文件的参数如下: rec.sph #负模聚类文件 1 #选择处理哪个簇 (<=0 为所有簇) N #以PDB文件的形式来生成表面 selected_cluster.pdb #输出文件的名 这是最一般的方法,选择的是最大的负模簇,也可以指定某处附近的簇,方法略。 三、生成栅格 1. 在活性位点周围生成一个盒子 输入命令:“showbox < box.in”,box.in文件也可以不写,程序会以问答方式进行。 2. 生成栅格 运行命令:“nohup grid -i grid.in -o grid.out>grid.log&”,运行这一步能够大大节省对接计算时间。我的grid.in文件在这里:UploadFiles/200 6-11/1120179826.rar Grid的参数以后再解释,不写在这里了。 四、刚性对接和柔性对接 刚性对接与柔性对接的不同只在于选择参数的不同,就不分开写了。命令都一样:“nohu p dock5 -i dock.in -o dock.out>dock.log&”,写不同的dock. in文件就行了。 我的刚性对接的dock.in文件:UploadFiles/2006-11/1120460197.rar;柔性
分子模拟 CDOCKER分子对接操作
HU J. 1 *此手册中底色为绿的为需提交的文件,其中截图放到word 文档中与其他文件一起打包提交至ftp 的相应文件夹。 I.对接计算软件: Discovery Studio Client v2.5.0.9164 (Copyright 2005-09, Accelrys Software Inc.) II.进入方法 开始菜单>Accelrys Discovery Studio 2.5>Discovery Studio Client III.设置路径(方便查找文件)Edit-Preferences- 以下操作涉及的文件均在Molecules 文件夹内 IV.文件准备 i.蛋白文件 将文件1AVD.pdb 拖入软件窗口,按ctrl+H 组合键,删除左边窗口的两个B 和Water 。第二个A 中删掉NAG600,只保留BTN400. (保留蛋白质A 链和配体A 中的BTN400.)
HU J. 2 Protocol :General Purpose | Prepare Protein 点图标栏绿色箭头运行,结束打开1AVD_prep.dsv ,保存为1AVD_prep.pdb 此窗口不用关 ii.对接小分子文件 把小分子文件BTN1_7.sdf 拖拽到新窗口。 优化分子结构作为对接初始构象:protocol :General Purpose | Prepare Ligand Input Ligands: BTN1_7: All Generate Tautomers: False Generate Isomers: False Lipinski Filter: False 其它参数默认 另存为BTN1_7.sd ,不要关闭子窗口。 V.活性部位的确定 切换到1AVD_prep 窗口; 定义受体:选中蛋白质A 链,变成黄色 Tools | Define and Edit Binding Site | Define Selected Molecule as Receptor ;系统视图中出现SBD_Receptor 。 定义活性部位:选中配体A 链,变成黄色 Tools | Define and Edit Binding Site | Define Sphere from Selection ;系统视图中出现SBD_Site_Sphere 。 调整活性球参数:在左侧选中球球(会变成黄金球球),在分子的3d 窗口右键attributes of SBD_Site_Sphere ,会显示有坐标和半径等参数(注意记录,需写入实验报告) 定义完口袋后,将原始配体 A 链删除。然后截图保存为
分子对接
分子对接技术简介 胡远东 1. Docking small molecules with LibDock tutorial 目的:给一组配体分子和蛋白活性部位,探索使用LibDock进行对接和分析 所需功能和模块:Discovery Studio Visualizer client, DS LibDock, 和DS Catalyst Conformation. 所需数据文件:pdb1kim_protH.msv和TK_xray_ligs.sd. 所需时间:20分钟 介绍 本教程中,一组配体分子将被对接到胸苷激酶(thymidine kinase)中,本教程包括: ?准备分子对接体系,执行分子对接计算 ?分析配体对接姿态 准备分子对接体系,执行分子对接计算 1.定义受体分子To define the protein as the receptor 在文件浏览器中找到数据文件pdb1kim_protH.msv,鼠标双击,该蛋白将在一个新的三维窗口中出现。在系统视图中,展开
AutoDock-分子对接步骤
Discovery studio: File -> New -> molecule window : 出现如下窗口,将所要处理的分子*.sdf文件拖入,Chemistry -> hydrogens -> dele 去掉所有氢,选中质子化的氮原子 选中质子化的氮原子,Chemistry -> charge:+1. Chemistry -> hydrogens -> add.(加上所有氢,包括质子化的氢原子) Pdbqt格式准备(pymol): 4M48-DA T.pdb用pymol读入:点右下方S键, 在左上方显示序列,选中21B,右侧显示有sele后,file -> save molecular: sele - OK -重命名ligand.pdb File -> open -> Duloxetine_3d.sdf Save > molecular : Duloxetine-3d.pdb 受体准备: 1. Pymol软件:pymol打开两个蛋白分子的pdb格式,将SERT-model叠合到4M48-DA T上,保存叠合后的SERT-model.pdb。(此步骤是重新定义SERT-model的坐标) 2.AutoDockTools: file -> ReadMolecule: SERT-moldel-align.pdb Edit -> Hydrogens : polar Only -> Ok Grid -> Macromolecule -> choose : SERT-moldel-align. 保存为:SERT-moldel-align.pdbqt 配体准备: Ligand -> input -> open : (*.pdb) ligand.pdb | Duloxetine-3d.pdb Ligand -> Output -> Save as PDBQT: 保存为*.PDBQT文件。 找活性位点,计算格点: Grid -> Macromolecule -> open : SERT_model_align.pdbqt 弹出对话框:NO、确定、确定Ligand -> input -> open :ligand.pdbqt 弹出对话框:确定 Grid -> set map types -> choose ligand : ligand Grid -> GridBox -> center -> center on ligand : 存图片或output grid dimensions file | close
史上最全药物研发过程全解
史上最全药物研发过程全解 我们身边常用的药物多少岁了? 青霉素:1941年,76岁;病毒灵:1960年,57岁,而最常备的感冒药阿司匹林于1898年上市,至今已有119年的历史了。这些老牌药物在科技发展如此迅速的今天,为何依然活跃于每个人的生活中? 药物研发之路有多难,看一看就知道了。(此处,小编回想到无数个泡在实验室的日日夜夜,已经哭晕在厕所) 一、结构筛选 结构筛选是新药研发过程中至关重要的一项,决定着项目的生死存亡。 1.药物靶点的确认 这个是所有工作的开始。只有确定了靶点,后续所有的工作才有展开的依据。确定靶标和靶标分析需要基于治疗的疾病,做大量的文献调研和生物信息分析,从基因序列到晶体结构,从基因组学到蛋白质组学。这一阶段整理的信息特别重要,将直接影响和指导新药研发的全流程。陶素生化可提供涵盖大部分已知通路和靶点的筛选工具库,助力药物靶点的确认,最受客户欢迎的工具筛选库包括激酶库,GPCR库等,通过这些工具小分子的对靶点活性和功能筛选,鉴定,可以大大加快靶点确认工作的进程。 当科学家发现某个家族或某类蛋白中的一个/几个蛋白对其感兴趣的生物现象的发生起了非常重要的作用,他们会用各类相关的小分子抑制剂,逐个打断其中的蛋白,观察是哪个蛋白的打断对该生物现象的 发生有明显的影响,从而确认其所感兴趣的哪个/些蛋白。 例如:科学家发现Rheb通过X蛋白调控自噬的发生。同时,通过研究发现蛋白是一个激酶。此时,科学家便可以用激酶的库进行筛选。通过筛选便可以简单确定X蛋白是哪个/类激酶。后续,通过RNA 干扰等基因水平的技术进一步确认X蛋白的作用等。
2.Hit的发现与获得 发现苗头化合物(hit),hit是指对待特定靶标或作用环节具有初步活性的化合物。发现hit的主要途径包括随机筛选的方法和理性设计的方法,理性设计的方法主要基于受体或配体结构和机制的分子设计。人工进行分子设计是一项复杂艰难,费时又烧钱的庞大工程。药物研发工作者很多时候采用虚拟筛选的方式获得hit。
天津大学网络教育学院计算机辅助药物设计期末复习总结资料.doc
计算机辅助药物设计 (请复习的同学参见各章节的总结视频) 名词解释 1.计算机辅助药物设计:用计算机处理并在屏幕上显示生物大分子和药物分子模型,特别是计算机辅助设计技术与合理药物设计过程的结合则称为计算机辅助药物设计。 2.手性药物是指药物分子结构中引入手性中心后,得到的一对互为实物与镜像的对映界构体。这些对映界构体的理化性质基本相似,仅仅是旋光性有所差别。 3.受体:可以识别和特异性地与具有生物活性的化学信号物质结合,从而激活或启动一系列牛物化学反应,产牛特定牛物效应的牛物大分子。 4.配体:能与受体产牛特异性结合的牛物活性分子,一般为小分子。 5?激动剂:能与受体结合并激动受体而产生其生物活性效应的物质或药物。既有亲和力又有内在活性的药物。 6.拮抗剂:能使受体结合,阻碍内源性物质与受体结合而导致其生物作用的抑制, 也称抑制剂。只有较强的亲和力,无内在活性的药物。 7.锁和钥匙学说:酶与底物结构上的互补性,用锁与钥匙的关系来描述酶与底物的关系。但此学说不能解释酶的逆反应。 8.速率学说:药物与受体相接触即产牛牛物效应,药物作用与其占有受体的速率成正比,而与其占有的多少无关。 9.占领学说:受体只有与药物结合才能被激活并产生效应,而效应的强度与被占领的受体数量成正比。 10?内在活性学说:药物与受体结合不仅需要亲和力,而且还需要有内在活性才能激动受体而产生效应。 11.二态模型学说:受体的构象分活化状态(R*)和失活状态(R)。两态处于动态平衡,可相互转变。 12.反义核酸:指人工构建的寡聚核昔酸,它能与特定mRNA精确互补、特异阻断其翻译的RM或DM分子,从而特异地封闭某些基因表达。 13?先导化合物:通过各种途径、方法或手段获得的,具有一定生物活性的新的结构类型化学物。活性不高、特异性差、副作用大、药代药动性质差。 14?先导化合物结构优化:对先导化合物做进一步的结构修饰和改造,提高活性和特异性,改善药代动力学特性,衍生出选择性高、安全性好。 15?药效基团:指一系列生物活性分子所共有的、对药物活性起决定作用的结构特征。 16.药动基团:指药物中参与体内药物的吸收、分布、代谢和排泄过程的基团。药动基团本身不具有显著的生物活性,只决定药物的药动学性质。 17.毒性基团在一些药物中,如果药效基团所产生的生物效应为毒性反应(毒性、致癌性、致突变性),这些基团称为毒性基团。 1&构效关系是指从定性的角度讨论药物的化学结构和生物活性的关系,即分子结构和构彖的变化与生物活性的有无及强弱之间的关系来推测药物的作用方式和机理。 19.生物电子等排:电子等排体指电子数目及排布情况相同的化合物或基团构成的电子等排体,它们具有相似的物理性质。在牛物领域里电子等排体表现为牛物电子等排。 20.药物潜伏化:利用药物代谢动力学知识,改变化合物的结构,从而改变其理化
同源模建的方法与结果分析
同源模建的方法与结果分析 Version 1、0、2------------------------------------------------------- 序言:作为一个以实验为主的生化工作者来说,很多时候可以通过分子生物学手段获取自己需要的目的基因,并在各种表达载体与宿主中进行对应蛋白的表达,随后对于这些蛋白的特性进行研究,这也就是一般酶学研究的特定套路。而近十几年来,人们开始思考就是否能够将特性与蛋白质的三级结构进行关联,从分子水平理解蛋白质与底物之间的相互作用呢?于就是类似于蛋白结构模建、分子对接、分子动力学模拟、量化计算等多种手段相继被创造以及应用。在这些方法中,同源模建无疑就是最基础也就是最重要的一个步骤,因为其质量的好坏直接决定了后续工作就是否可信。因此,本文打算就同源模建的基本原理、常用软件及服务器以及结果分析与改进提供一些个人的经验,并希望各位朋友能够给予批评指正。 1.同源模建的原理及应用限制 两点基本原理: 1、一个蛋白质的结构由其氨基酸序列唯一的决定。知道其一级序列,至少在理论上足以 获取其结构 2、结构在进化中更稳定,变化比序列层面的变化要缓慢许多。 应用限制:模板蛋白与目标蛋白的序列一致性需要大于30%,且越大建模准确性越有保障。 了解了基本的原理,我们需要知道在实际操作中,同源模建都需要怎么样进行。 同源模建的过程从实践中可分为以下7个步骤: 1.模板识别与初始比对 在序列一致性比较高的时候,可以通过简单的序列比对程序如BLAST获取目标蛋白 的结构(将比对的数据库选择为PDB数据库)。 2.比对结果的校正 用以上的方法确定一个或多个建模模板后,应该采用更为精确的方法已取得更优的 比对结果。有时在序列一致性较低的区域比对两条序列可能会具有困难,这个时候, 我们可以采取其她同源蛋白序列一起参与比对来找到解决的办法。 3.主链生成 比对完成后,就可以开始实际的建模过程了,相对与后面几步来说,主链建模时最没有 难度的一步了,因为大部分软件都就是通过简单的拷贝模板蛋白的主链坐标来实现 这一目的的。 4.环区建模 这一部分主要就是目标蛋白与模板蛋白的比对结果中存在缺口的部分如何处理的 问题。第一种解决的方式就是略去模板蛋白存在的残基,留下一个必须补上的缺口。 另一种情况就是将主链截断,插入缺少的残基。 5.侧链建模 当我们比较结构相似的蛋白质中保守残基的侧链构象时,我们会发现她们的侧链构 象通常会比较相似。这就告诉我们如果加保守残基的侧链构象完整的拷贝到模建蛋 白上时,在某些时候比先拷贝主链构象之后,再预测侧链构象来的可靠。但就是这一 经验规则在实际运用中仅在两者序列一致性较高,并且保守残基之间形成接触的情 况下才能实现。因此,在现有的测序中,都就是构造各种可能的构象体,并利用基于能 量的函数打分来实现侧链构象的选择的。 6.模型优化