Magix X射线荧光光谱仪分析方法的制定步骤

MagiX X射线荧光光谱仪分析方法的制定步骤
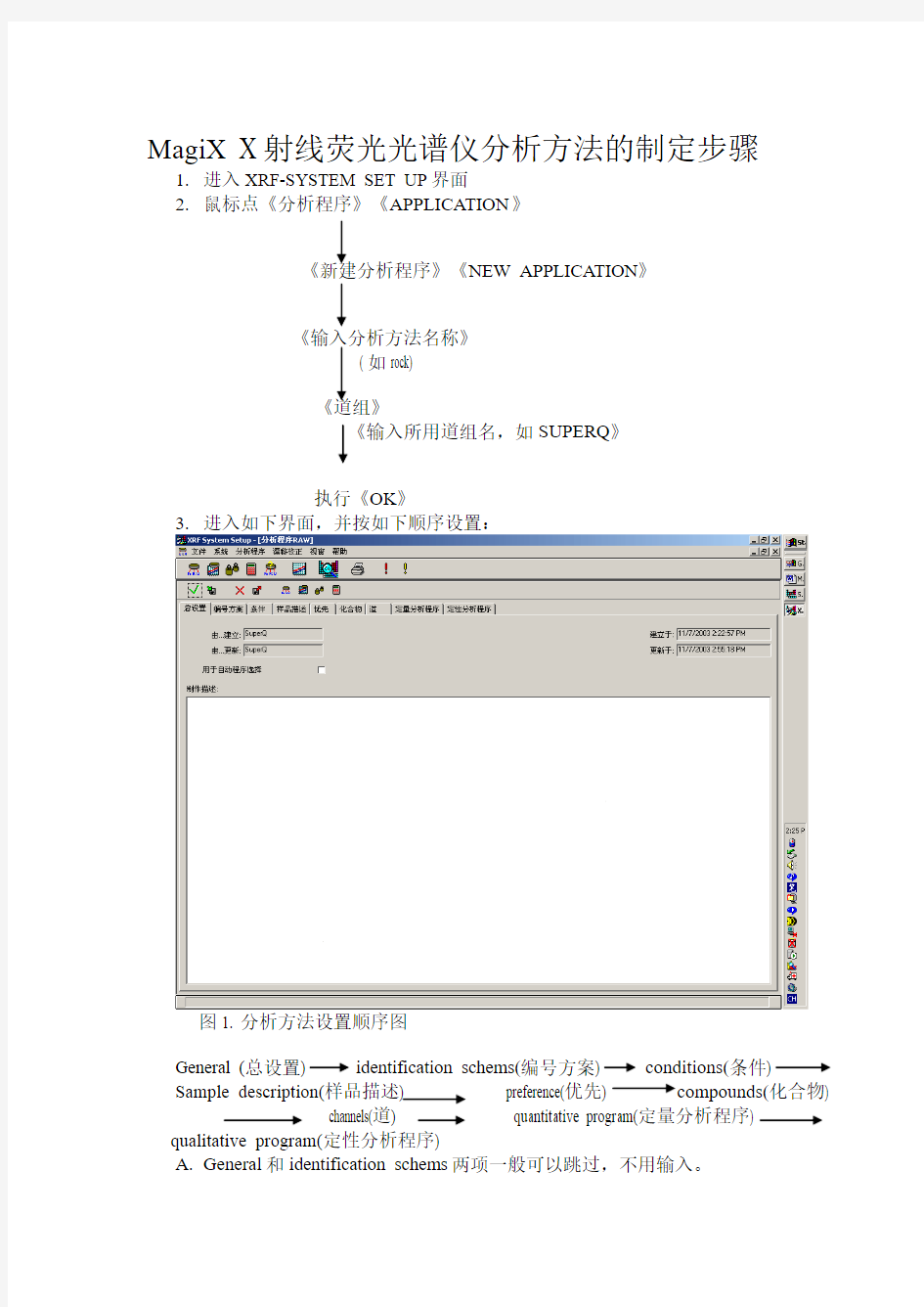
1.进入XRF-SYSTEM SET UP界面
2.鼠标点《分析程序》《APPLICATION》
《新建分析程序》《NEW APPLICATION》
《输入分析方法名称》
( 如rock)
《道组》
《输入所用道组名,如SUPERQ》
执行《OK》
3.进入如下界面,并按如下顺序设置:
图1. 分析方法设置顺序图
General (总设置) identification schems(编号方案) conditions(条件)
Sample description(样品描述) preference(优先) compounds(化合物) channels(道) quantitative program(定量分析程序)
qualitative program(定性分析程序)
A.General和identification schems两项一般可以跳过,不用输入。
B.鼠标点conditions(条件):XRF-vacuum lock time(s)(XRF真空锁定时间,系指样品杯放进样品室中,盖上盖板后预抽真空时间,粉末压片如水泥工业中生料可设定10秒,而钢铁或熔融片通常设4秒即可)。delay time(s)(延迟时间,系指试样进入测量位置后再抽真空的时间,通常设置2秒)。Spinner on(自旋),通常没定自旋,以防止样品不均匀。Measure in decreasing energy order,该项通常按能量从大到小进行测量,鼠标在方框中点一下,但若需测定氯或硫时,即不选用该项,应先测氯或硫。
图2.分析方法中‘条件’框图
分析光路介质,通常选真空(Vacum),液体样品选氦气(Heleum)。狭缝罩具的选择取决于样品杯内孔径大小。
C.鼠标点样品描述(sample),根据试样的物理状态可选用熔融块 (bead)、液体(liquid)、粉末压片 (presed power)、固体 (solid)等状态中的一种。选用熔融块(bead)、液体 (liquid)、粉末压片 (presed power)、)时,要输入样品重量、熔剂、或溶剂或粘结剂或助磨剂(硬脂酸的分子式为C18H36O2)的重量。
选粉末压片法,若以20.0克样与助磨剂0.20克硬脂酸为例,可参见下图3。
选熔融样品时,烧失量有三种情况可选择:No LOI,Manual input和Balance。选No LOI,表明不存在烧失量,选Manual input,表明在测量标样或样品时,需输入烧失量,选Balance时,则用100%-所测氧化物的总和。样品重量和熔剂重量可有两种形式输入,即选或不选固定比例,选固定比例只要选一次,但每个标样和样品的重量和熔剂的重量是相同的,至少比例关系是固定的。若不选固定比例则每个标样和样品的重量和熔剂的重量根据实际称量输入。
需要指出的是以何种物理形态样品进行测量,需依据对分析方法所要求的准确度和精度、分析时间、标样等因素确定。
图3.分析方法中‘样品描述’框图之一……粉末压片法
D.鼠标点compounds(化合物) ,从add compound化合物表中加入所要分析的化合物和率值,如Al2O3,SiO2……KH等加进去。若分析以元素表达,也可从周期表中直接点所要分析的元素。无论以何种方法输入,总是按分析报告的顺序加入。
图4.分析方法中‘化合物’框图
E.鼠标点channels(道) ,通常化合物选定后,进入道可以见到软件给出的参数。
图5.分析方法中‘道’条件的选择
但要根据样品实际情况,选择所用分析线(line),如Kα、Lα,kV和mA,晶体和滤光片,选角度angle(2θ),测量,显示谱图,其虚线为理论角度或先前己测角度,实线为现测得的角度,角度确定后在谱一侧或两侧平坦处扣除背景,然后输入待测样待测元素的浓度,并依据浓度确定CSE%或LLD,将其销定,计算峰和背景测定时
间.
鼠标点PHD(脉冲高度分析器),再测量决定上阈和下阈值。
上述条件的选择可参见附录1。
F.鼠标点quantitative program(定量分析程序),最后确定分析时间。存盘后退出。
G.标样浓度(在界面上以两个天平法码表示)栏,选择分析方法(RAW)名称,将标样名称加入,并输入标样各组分浓度。
H.将分析方法先存盘,再在SYSTEM SET UP中点漂移校正,建立新的漂移校正,如RAW,具体方法参见附录3。
I.打开分析方法,在MONITOR一档中选择上面所确定的漂移校正方法名称。
4.进入XRF-MEASURE & ANALYSE界面,选择测量(MEASURE),再用鼠标点标样测定栏(以两个天平法码表示),然后鼠标点分析方法(rock)名称,即在右侧显示标样名,将待测标样放进试样室,鼠标点标样名后,再点MEASURE。按此将全部标样测完。
5.再进入XRF-SYSTEM SET UP界面,在《APPLICATION》栏中打开校正曲线计算程序(或用鼠标点计算器形象图),用鼠标选定分析方法(RAW)名称,在出现的界面上,在待测元素上方,用鼠标点之,使待测元素档呈深色,再用鼠标点下方的计算(CALCULATE),检查每个待分析元素的曲线,每个标样的计算结果和化学值之间差是否满足要求。若某元素不满足要求,如Si。则在本计算界面的下方,鼠标点Si 后,Si档呈深色,再点ADD,从 栏中点Al,OK后再计算,以此类推。
亦可用理论α系数,即在下方用鼠标点correction coefficients下方所要计算的元素,再点理论(theory),进入理论α系数计算界面,在calculate alphas上用鼠标点之,然后执行OK。
粉末压片法较少使用理论α系数法和基本参数法,即使用也要适当加经验系数,校正颗粒度和矿物效应。熔融法和金属样品一般用理论α系数法和基本参数法,有时为了校正因加工工艺等因素引起的结构效应,也适当引用经验系数,如NiFeCo程序中γ系数。
6.将 matrix model中CLASSIC改为基本参数法(FP),用鼠标点Calculate。
7.经α校正后,若某标样仍超差,可在曲线中删去,方法是用鼠标点超差的标样
后,再用鼠标点曲线下方的(EXCLUDE)。若欲恢复,则用鼠标点欲恢复的标样,然后按下方的包括(INCLUDE)。但是标样含量中最低和最高的标样,最好不要删去。
对删去的国家标样,需要查明原因,是含量输错还是制样引起的。
附录1波长色散谱仪定量分析条件的选择
1 X光管的高压和电流的选择
端窗靶X射线管,高压电源供给X射线管的电压和电流为60kV和125mA或更高。设置的X射线管高压和电流的乘积不能超过谱仪给出的总功率,MagiX系列谱仪通常有2.4、3和4 千瓦之分,,2.4kW的X射线管的电压和电流为60kV和100mA。
SuperQ软件所推荐的电压和电流,其乘积通常是满功率,而且电压和电流值随元素而异。推荐值是依据元素而定,并未根据实际试样。众所周知,在选择高压时,其必要条件是设定的值必须大于待测元素激发电位,当所加电压是待测元素的激发电位4倍时,通常可获得90%的强度。管流可根据试样中待测元素含量进行选择。高压推荐的一般原则列于表1。长期实践表明,在许多情况下不应采用满功率,而应取满功率的
0.7 ~0.9倍,这有助于延长X 射线管的使用寿命。此外,若测定多个元素,其管压和管流的选择也不能变化太多,通常只需设定两种,如30、60kV 分别满足轻、重元素的测定即可。这样有助于提高工作效率和仪器的稳定性。对于水泥工业中生料、熟料中诸成分的分析,用MagiX 谱仪通常采用50kV ,40mA 。 2 角度的校正、背景的扣除和计数时间的确定
角度的选择取决于待测元素所选的谱线和晶体,当这两个条件一旦定下来后,软件将提供2θ角度值等。
A.谱线的选择原则
谱线的选择大体遵循下述原则:
(1)通常选择αβαβαM L L K K 和、、、等几条主要特征谱线,这些谱线在各自谱线系列中是强线。对同一元素,其灵敏度依次为αββααM K L L K 和,,,。通常选择ααL K ,线。 (2)选择何种谱线视试样和制样方法而异。如元素Zr 若含量较高(>20%),用熔融法制样时,为避免厚度的影响则选用αL 线,而不是αK 。这主要由于X 射线荧光透射试
样的厚度与射线的能量和基体成函数关系,令有99%的X 射线荧光能透射到试样表面,其强度以d I 表示,0I 为样品表面的强度,则
2)(0
99.0ψμρSin e I I d d
*==- (1) 2)
99.0ln()(ψρμμSin m d **-=
(1)式中μ为基体对X 射线荧光总质量吸收系数,ρ为基体的密度。为了说明问题,现将不同谱线在不同基体的透射厚度列于表2(使用PW24系列谱仪)。。
表1 高压的选择
由于待测元素和基体对原级X 射线谱和待分析元素的特征X 射线的质量吸收系数存在很大差异,待测元素相对强度和浓度之间的关系,并不呈比例。如表3中列出的铁及其各种氧化物中FeK α谱线相对强度及临界厚度数据。
从表3可知,当试样中铁的浓度从70%增加到77.8%,而FeK α线的相对强度仅从95%增加到95.7%。究其原因是氧对原级谱和FeK α谱基本上是透明的,即吸收系数很小。而基体中铁基本决定各种氧化物的吸收系数、临界厚度以及试样中FeK α线的出射厚度,由于在70% Fe 2O 3中FeK α临界厚度增大,因此其相对强度依然较大。
表3 铁及其氧化物中FeK α强相对强度和临厚度数据
(3)在可能的情况下,所选谱线应避免基
体中其它元素的谱线干扰。在X 射线荧光光谱分析中,特别是波长色散谱仪,与其他发射光谱相比,其干扰是很少的,但在有些情况下谱线干扰不仅存在且很严重,如PbL α和AsK α谱线几乎完全重叠,因此为获得待测元素的净强度,必须对谱线干扰进行校正。现代谱仪的软件通常可提示或自动予以校正干扰谱线。干扰谱线校正的方法通常是:
(a).如在测定低含量As 时,一般不选用AsK β线,若Pb 的含量与As 相比较,不是很高时,可选AsK α用于测定砷;选PbL β测定Pb ,然后在仪器提供的软件中,用数学方法扣除铅对砷的干扰;
(b).此外,测定超轻元素时,对试样中可能存在的元素高次线或L和M线对其干扰应
予注意。在制定分析方法时,因元素的谱线在不同化学状态下有少许差异,故需要用实际样品经扫描确认2θ角度。
(c).谱线的干扰还应考虑间接干扰谱线的干扰。如测定Zr时,Nb对Zr有干扰,而
Mo和Y对Nb又有干扰,因此,在测Zr时不仅要考虑Nb的干扰,还应考虑Y和Mo的间接干扰。为便于了解谱线干扰,现将常见的谱线干扰列于表1。
谱线干扰也可通过选择狭缝和分辨率高的晶体予以避免。如用LiF220代替LiF200、细狭缝代替粗狭缝减少干扰,但要损失强度,这需要加以权衡。常见的谱线干扰参见表4。在通常情况下,不仅要考虑直接干扰的谱线,也要考虑间接干扰元素。
若在待测元素中,未考虑干扰元素,应在道设置中考虑之,如测硫时应考虑钼的干扰,虽然标样中无钼的元素。
B. 背景的扣除
背景产生的原因一般可分为两类,其一是由样品引起的,原级X射线谱在样品中产生散射(康普吨散射和瑞利散射)线,其强度随样品成分的变化而发生变化;此外在被测谱线附近存在谱线干扰,这种干扰谱线来源于原级谱散射线或样品本身;其二是由样品产生的射线与仪器相互作用造成的背景,如晶体荧光和分光晶体引起的高次线等;此外宇宙射线和样品中放射性核素和电子学线路也能产生背景。背景对微量元素的检测限和准确度均有较大影晌,因此,通常均给以足够重视。
经过多年的研究,背景校正已发展成如下方法:理论背景校正法、实测背景扣除法、康普吨散射校正法、经验公式校正法。在这几种方法中,实测背景校正法效果最好,对上述两种背景均可进行修正,而理论背景校正法、康普吨散射背景校正法仅能修正原级谱在样品中的散射引起的背景,理论背景的计算目前尚不能用于实际工作。实测背景的选择原则:对于主次量元素,即峰背比在50以上时,若谱峰对称,仅需要选谱图尾部一点作背景;若非对称谱,或有谱线干扰,通常需选2个背景点。
在许多情况下,常选用公用背景,作为测量多个元素背景,这样做不仅缩短了测量时间,还可克服在某些分析线附近不易找背景点的困难,此外,公用背景点可用作内标,公用背景的选择与分析试样组成和所测元素有关。如在分析化探样品中 Nb 、Zr 、Y 、Pb 、Rb 、U 、Th 和 Bi 等元素时,用LiF 200时可选用24.55度为公共背景点,42.65度为Ni 、Cu 、Zn 和 Co 的公用背景点;在用熔融法分析氧化物硅酸盐和碳酸盐时,在测 定Al 、Si 、P 、 S 的K α线和Zr 、Y 的L α线时用0.7590nm 为公用背景点;分析铁基镍基钴基合金中Ta 、Hf 、Re 、W 的L α 线和Ni 、Co 、Fe 、CR 的 K α线分别用0.1407nm 和 0.2039nm 为 公 用 背 景。背景的正确扣除可以有效的降低检测下限。
C.测量时间的确定
现在是根据对计数统计误差的要求或对检出限的要求确定测定时间,测定时间是可变的。
根据Jenkins 等指出,X 射线荧光分析的标准偏差与被分析成分的浓度具有如下关系:
C K S XRF = (2) K 为精密度品质因子,C 为浓度%,K 值一般在0.01~0.05之间,在此基础上,可用下式计算:
%100005.0%*=
C
C
CSE REL (3) CSE 为计数统计误差,将浓度及CSE 输入后,即可求得峰位和背景的测定时间,若分析精度要求不像钢铁样那么高,也可适当放宽。(6)式的前题条件,测定时间的设置以测定100%含量试样时,在所设测定时间内可收集不少于4×106计数。浓度与REL CSE %和
误差关系参见表5。
在校正角度时,CSE 是以浓度为单位,而不是计数率(kcps),灵敏度(kcps/%)定义为:
C
P P S B
P -= (4)
对于(5)式可改写成:
S
kcps CSE CSE )
()(=浓度 (5)
然后给出:
b T R
S LLD *=3000 (6)
式中R 是己扣除背景后的计数率,以kcps 表示,b T 为背景测定时间。亦可根据待测试样对检测限要求确定背景和峰位计数时间。其公式: b
b
T R S
LLD 23=
(7)
b R S 和分别是灵敏度(cps/%)和背景的强度。
在净强度大于等32倍的背景强度计数标准偏差时,测定时间的延长,可以降低检测限,在同样条件下用PW2400测定20个元素,测定时间分别为10分钟和60分钟,其结果如图2所示。
表6 浓度与
3 脉冲高度分析器
使用脉冲高度分析器主要目的是处理探测器给出的电脉冲信号,由于晶体在衍射时可能产生晶体荧光、某些元素的高次线或高次线的逃逸峰、待测元素本身产生的逃逸峰,这些信号所产生的电脉冲如不处理,均影响分析结果的准确度。
MagiX 系列谱仪提供了O-255道多道脉冲高度分析器,可选用两个不同的PHD 值,是十分有利的,它不仅能将四级线显示,还可提高计数率。 A.逃逸峰的处理
图2.在同样条件下,测定时间对检测限结果的影响
逃逸峰的产生,以流气正比计数管为例予以说明。从晶体衍射出来的X 射线荧光,其能量大于等于Ar 气的K 层电子激发电位时,可使Ar 气产生ArK α线,Ar 气对
Ar αK 吸收的可能性很小,就是说Ar αK 线能量从探测器逸出而损失。如待测元素的能量为Ei,Ar αK 能量为2.96keV ,则逃逸峰的能量为两者之差,即Ei -2.96keV 表示。以Cr αK 线为例,Cr αK 能量为5.41keV ,逃逸峰的能量等于5.41keV -2.96Kev=2.45keV ,待测元素的PHD 的峰位通常锁定在50%的位置上,对Cr αK 而言,5.41 keV 相应于50%,那么逃逸峰的位置:
%7.2250
41
.545.2=* 因此在PHD 分布曲线中可见两个峰,如图3所示。
基于每个待测元素的脉冲高度通过微机均锁定在50左右,因此逃逸峰在PHD 中的位置是与待测元素的能量有关,能量逾大,则逃逸峰的能量也大,其在PHD 中位置则逾靠近主峰,如Cu αK 的逃逸峰的能量为5.08keV ,在PHD 中位置为 31.5外,而 K K α 的 逃 逸 峰 的 能 量 为 0.35 Kev ,在 PHD 中 位 置 是 5.3。
逃逸峰是否要包括在PHD 下阈和上阈之间,视待测元素含量而定,通常含量较低时,其逃逸峰不包括在内,反之则需包括在内。
图3 Cr αK 谱线及其逃逸峰的PHD 图
B.利用PHD 消除晶体荧光的影响
来之于样品的X 射线荧光照射到晶体上,晶体中某些元素产生X 射线荧光,与待测元素X 荧光一起进入探测器。为消除晶体荧光,基于待测元素和晶体荧光的能量存在差异,故可利用PHD 予以消除。图4列出了Ge 晶体产生的GeL α线对P K α的干扰,并利用PHD 消除其干扰的情况。
图4 P 的 K α和 Ge L α 晶 体 荧 光 的 PHD 图
C.利用PHD 消除高次线的干扰
当基体中某一元素含量较高时,在其波长N 倍处亦会产生相应的谱,若待测元素的波长也在该处或附近,利用晶体是不能将它们分开。但这两个元素的谱线的能量 相差n倍,故可用PHD 将它们分开。
在图5中列出PX1晶体测石灰石中Mg 的PHD 图,从图中有四个峰:〈1〉、在 50%处MgK α峰;〈2〉、CaK α的三级线峰位在3×50=150%处;〈3〉、CaK α的逃逸峰在30%处;〈4〉、来之于晶体PX1的晶体荧光,PX1是多层薄膜叠加而成,由W 和Si 组成,WM α也干扰MgK α测定,WM α峰位和Si K α峰位分别在:
8.7050253.1775
.150=*=*ααMgK WM E E
4.6950253.1739
.150=*=*ααMgK SiK E E
很明显,这两个峰是分不开的。在这种情况下,对PHD 下阈和上阈要仔细选择 。
D.利用设置两个PHD 上、下阀消除高次线逃逸峰的干扰
在分析重晶石中Al 时,BaL α 三级线的逃逸峰的能量是(4.465keV -2.957keV )= 1.50keV,与AlK α峰能量完全复盖在一起,如图6所示。
MagiX 谱仪可设置两个PHD 予以消除,具体办法 : AlK α纯强度=窗口1(AlK α (gross)+BaL α逃逸峰)-F×窗口2(BaL α谱),因 子F 可用不含Al 的BaCO 3计算出来。 当然这种类型的干扰也可在基体校正中利用谱线干扰予以消除。
此外,当流气和封闭式正比计数管联合使用时,测定较重元素时如CuK α,在PHD 图上可显示Ar 和Xe 两个逃逸峰。如图7。
图 5 PX1 用于测定石灰石中 Mg 的 PHD
图6 测定重晶石中 Al2O3 的脉冲高度分布图
图7 Cu K 逃逸峰的脉冲高度分布图
附录2 校正曲线的制定
定量分析是以一组标样中待测元素的强度和浓度值,通过最小二乘法或作图法求得待测元素的强度和浓度之间的关系。在可能的情况下,要求强度是净强度,即已扣除背景和干扰谱线后的强度。
进行定量分析时,一般需要制定校正曲线,将测得标样的强度与其含量建立如下的关系:
]1[i i i i i i M R E D LO C +**++= (1) ]1)[(i i i i i i M R E D LO C +*++= (2) ]1)[(2i i i i i i i i M R F R E D LO C +*+*++= (3)
(1)式应用于背景校正后的情况,(2)式用于不校正背景的情况,(3)式只用于特殊的情况,如分析从线性到中间厚度的样品,可以二次曲线表示。LO 是谱线重叠校正,可以用干扰谱线的强度或浓度进行校正。其实在实际工作中,D 和E 是通过回归方程计算一组标准样品获得的,并且是常数。在方程(1)中截距i i i E Rb D *-=,强度截距i Rb 是没有从一组标准样中测得的强度i R 中减去残留背景的计算平均值,而不是单独测定的背景强度,严格的讲,只有i i Rb D 和为零,i R 方是纯强度,否则它就包含有残留背景,并且参与基体校正,这有可能
引入误差。
基体简单,不要考虑元素间吸收增强效应的影响,即M 为零,根据标样的含量和测得的强度用回归法直接计算D 和E 。或在方格纸上作校正曲线,其横坐标以含量表示,纵坐标为待测元素的强度。在测定低含量元素时,为消除基体效应,亦可将待测元素的强度与内标元素强度比或与散射线强度比用你纵坐标。曲线截距D 的单位为含量,斜率E 的倒数为方法的灵敏度,E 的单位为%/kcps 。为了获得准确的结果,亦可令D 值为零。
若在制定校正曲线时,基体存在较严重的元素间吸收增强效应,则需要校正。基于Philips 公司生产的PW14××和24××系列的仪器提出的PH 模式将常用几种校正模式合并为一个方程,为方便起见,引用如下:
∑∑∑∑====**+*+*+*+=+n j n j n j n k k j k j i i
ij j
ij j ij i C C C C C M 1111,,11]1[γδβα (4)
(4)式中C 是浓度或计数率, n 是待分析元素数,α、 β、δ、?是用于基体校正的因子。
i 是待测元素,j,k 是干扰元素。该式的特点有:
(a) 式中α因子可用De Joung 理论影响系数法计算,也可用经验方法计算。β,δ,γ为经验系数,根据标样的浓度和强度通过数学上解方程求得;
(b) 理论α系数的计算可用基本参数法根据标样各待测元素的平均含量求得,当然可通过设置二元或三元组分后,用基本参数法计算,PH 模式是用平均含量计算理论α系数的;
(c) (4)式将理论影响系数和经验系数相结合 ,强度校正和浓度校正相结合,为使用者提供方便,可获得质量更好的校正曲线。这样做,不仅可以校正元素间相互影响,还可对粉末压片法中对矿物效应和颗粒度效应进行部份校正;
(d) (4)式将L-T 方程或De Joung 方程、R-H 方程、C-Q 方程合成于一个方程,可根据实际情况予以选择;
(e) 在软件设计上,可对干扰系数、影响系数单个独立计算, 计算后将该系数锁定,再计算另一系数,这样可用少量标样获得多个参数可靠值。
这里需要指出,在一般情况下不论用何种方法迸行基体校正, 应遵循下述诸点: (a) 标样的浓度范围需复盖所测元素的浓度 ;
(b) 校正曲线中间部分所计算的浓度比其两端更准确 ;
(c)在同样条件,用多个标样获得的校正曲线分析未知样比用单个标样更准确。
(d)标样与未知样的物理化学状态一致和制样方法一致是获得准确结果的必要条件。
图.理论α系数校正结果
基本参数法简介
随着计算术技术迅速发展,基本参数法已用于常规试样分析。基于计算较复杂,这里仅作简单介绍。 1 基本参数法原理
依据X 射线荧光强度的理论公式(1),公式中所用的几何因子、各种基本参数(荧光产额、质量吸收系数、吸收限跃迁因子和谱线分数)、晶体衍射效率和探测器的探测效率均准确的情况下,可以通过测得试样中诸组分的纯强度,计算出试样中的各组分的含量。上述基本参数存在一定的误差,晶体衍射效率和探测器的探测效率均随谱仪不同而有所不同。因此,目前基本参数法仍然需要通过标准样品的组成含量计算出理论强度,并将各组分的理论强度和实际测量的强度进行比较后,方可对试样进行分析。
若仅考虑基体的吸收效应,即原级谱对试样的激发,其表达式为一次X 射线荧光,可写为:
P i = ∑**?****i abs s
i i i i I D C G ,min
,,λλλ
λλμμλ (1)
原级X 射线光谱波长在短波限(λmin )和待测元素和基体元素的吸收限(λabs )之间。 若考虑增强效应,则需计算二次荧光和三次荧光。在一般情况下只考虑二次荧光,其公式写为:
S ij = )(,,min
,,j ij j
abs s i j i
i C e I D C G ***?***∑*
λλλ
λ
λλμμλ
(2)
在上述式中引入D i, λ和D J,λ。数值分别是零和1,原级X 射线光谱波长在短波限(λmin )
和待测元素和基体元素的吸收限(λabs )之间,D i, λ为1,否则为零。同样,当基体元素j 的波长大于待测元素的吸收限时,D J,λ为零,反之小于待测元素的吸收限时,D J,λ为1。
因此通常用基本参数法校正元素间吸收增强效应时,既要考虑吸收也要考虑增强效应,应将一次荧光和二次荧光一并考虑。
I i =P i +S ij (3)
式中符号的物理意义参见”X 射线荧光光谱分析”一书第二章。 2 基本参数法的计算方法
基本参数法的计算过程很复杂,需要准确知道各种物理参数, 即原级X 射线光谱强度分布、 质量吸收系数、荧光产额、吸收限跃迁因子、谱线分数等,这些物理参数的物理意义及数据可参见第一章中有关内容及附录。Ωi 为几何因子,由仪器厂家给出。
将λmin 到λabs 之间的原级X 射线光谱划分成△λ基本单元,为达到求和的目的,可任意划分;原级X 射线谱强度数据最早用实测的谱线分布数据,现在常用在第一章中所介绍的Pella 和丰梁垣在实验基础上建立起来的公式[1-6,7]。但需要指出,在应用该公式计算原级谱强度分布的计算中连续谱和特征X 射线光谱所取的单位是不相同的。连续谱本身是一种分布函数(光子数随波长而分布),使用的单位:光子数/e-/Str./?,即在单位电子(e-)轰击下,在单位立体角(Str.)内和单位波长间隔(1?)内所产生的光子数。而特征X 射线光谱是一非分布数值,其波长只对应于某一特定的波长。所以它的单位应该是光子数/e-/Str.,即在单位电子轰击下在单位立体角内所产生的特征X 射线的数目。理论上特征X 射线光谱的自然宽度是几个电子伏特,可忽略不计。但用实验方法测得X 射线光谱谱线的宽度远大于此值,这是由于色散和探测系统的色散和分辨率的不足所造成的。故实际处理时应将整个特征谱线分布范围内的谱线面积作为特征X 射线波长处的强度。因此,连续谱和特征谱的量纲差一个1/ ?因子,而I 0(λK )ΔλK 的量纲则与特征谱相同。所以在原级X 射线光谱分布中考虑特征谱的方法是直接将它的强度加到离它最近的I 0(λ)ΔλK 中去,而不应该加到I 0(λK )中去后再乘上ΔλK ,否则引入严重的计算误差。基于基本参数法计算中通常仍用标准样品,并以相对强度为依据的,所以I 0的绝对值是不重要,可以等倍率将它放大或缩小,但不可改变其分布的形状和趋势。由于同样的原因,X 射线管的电流值与基本参数法的计算无关,因为管电流的变化不改变原级X 射线光谱分布的形状,而只改变其绝对值。
基于这些物理参数及几何因子的数值均有不可忽略误差,因此要进行强度I i 和含量C i 的绝对计算是很难获得理想结果的,解决的办法是将X 射线的强度表示成相对强度:
R i =)(i i I I
(4)
式中 I (i)为纯元素I 在与未知样同样条件下测定的强度。这样可消除参数误差和一些不确定因素对分析结果的影响。但在许多情况下获得纯元素的标准样有困难,而且在高电压和高电流情况下测定纯元素时,强度太高,有时可能超过探测器所允许的计数范围,很难获得纯元素的强度值。因此,通常用含有多个元素的标准样品代替纯元素的标准样,并用下述方法求得相对强度:
R i =)(i i
I I = (),(s i i I I )ms ×( )(),(i s i I I )cal (5)
式中, I i 为未知样中I 元素的强度, I (i)表示I 元素纯元素的强度,由计算获得,I i,s 表示
标准样中i 元素的强度。下标ms 表示实测值,cal 表示计算值。所谓计算值是根据标准样中i 元素和其它基体元素的己知含量从基本参数法公式计算出来的结果。
欲从相对强度计算待测试样中组分的含量,就需要用迭代法。迭代的过程以下: (1) 将测得的或从(6-5)式计算得到的试样相对强度(记为m i R ),作为各组分浓度的初始估计值C i 。
(2) 将C i 代入(6-3)式,算出各组分的相对强度(计算值,记为R i )。
(3) 将理论计算的相对强度值 cal i R 与测得的相对强度值m i R 进行比较,按下式对前一次的浓度估计值进行修正:C i ’=( m i R /cal i R )× C i ,式中 C i 为前一次估计值, C i ’为修正
值。
(4) 将修正后的浓度值 C i ’,进行再比较,再修正……,如此循环直到C i ’收敛为止(可先规定一收敛标准,如设前后二次算出的浓度值相差不超过0.005%,但所有组分都需达到此标准)。
为加速收敛,对C i 的修正通常使用双曲线三点内插法,即令
)1()()1(,,
i cal i cal i i m i cal i i m i C R R C R R C R C -+--= (6)
C i ’是从(0,0),(C i , cal i R )和(1,1)三点所限定的双曲线内插求得的。一般只需要3~4次迭代就可以达到所需的精密度。但计算结果的准确度与强度测量以及所用基本参数的准确度有关。
为便于了解计算过程,现将无限厚试样的基本参数法计算流程框图,列于图。
Nicoletis5红外光谱仪检定标准操作规程.docx
1目的 建立 Nicoletis5 红外光谱仪的检定规程,确保仪器的性能可靠和测量的准确性。 2范围 适用于 Nicoletis5 红外光谱仪的检定。 3职责 质量检验部负责执行此规程,质量保证部负责监督实施。 4定义 无。 5内容 5.1 检定项目和技术要求 序号检定项目技术要求 1波数示值误差在 3000cm-1附近的波数视值误差±5cm-1在 1000cm-1附近的波数视值误差±1cm-1 -1 附近的波数重复性 -1 2波数重复性在 3000cm≤2.5cm -1 附近的波数重复性 -1在 1000cm≤0.5cm 3透射比重复性不大于 0.5% 在 3200cm-1~2800cm-1分辨峰7 个 4分辨力2851cm-1与 2870cm-1之间分辨深度≥18% 1583cm-1与 1589cm-1之间分辨深度≥12% -1 峰半高宽 -1水汽 1554.4cm≤2cm 5本底光谱能量分布不小于20% 3200cm-1~2800cm-1内 100%线的平直度≤1% 6 线的平直度-1-1内 100%线的平直度≤1% 100%2200cm~1900cm 800cm-1~500cm-1内 100%线的平直度≤4% 7噪声不大于 1% 5.2 检定环境 5.2.1 环境温度: 16~25℃,相对湿度:≤60%; 5.2.2 仪器应置于平稳的工作台上,不应有强光、强气流、强烈振动和强电磁干扰;5.2.3 环境无腐蚀性气体、烟尘干扰;供电电源电压220V±22V ,频率 50Hz±1Hz。 5.3 标准物质
聚苯乙烯膜红外波长标准物质。 5.4 检定内容 5.4.1 波数示值误差与波数重复性 Nicoletis5 红外光谱仪扫描范围为4000cm-1~400cm-1,分辨率为 4.0cm-1,常用扫描速度,扫描次数为 15.待 Nicoletis5 红外光谱仪稳定后,采集空气本底背景,扫描聚苯乙烯红外波长 标准物质,测量 3027cm-1, 2851cm-1,1601cm-1,1028cm-1,907cm-15 个主要吸收 峰。重复测量 3 次。按公式( 1)计算,取△V绝对值最大值为波数示值误差。按公式(2)计算,取δv绝对值最大值为波数重复性。 v=v i- v(1) δv=v max-v min(2) 式中: v——波数示值误差, cm-1; δ v——波数重复性, cm-1; -1 vi ——第 i 峰值波数测量平均值, cm ; -1 v max——第 i 峰波数测量最大值, cm-1; -1 v min——第 i 峰波数测量最小值, cm 。 在 T 绝对值最大值为透射比重复性。 R T=T max-T min( 3) 式中: R T——透射比重复性, %; T max——聚苯乙烯峰值透射比最大值,%; T min——聚苯乙烯峰值透射比最小值,%; 5.4.3 分辨力 分辨苯环特征吸收峰的个数 -1~2800cm-1范围内,谱图可分辨的吸收峰的个数,见图1。 分辨深度 -1(峰)与 2870cm-1(谷)之间的峰谷深度和1583cm-1(峰)和 1589cm-1(谷)之间的峰谷深度,用T 表示。见图 2 和图 3。 5.4.3.3 半高宽
X射线荧光光谱仪介绍
X-射线荧光光谱仪(XRF) 1、仪器介绍 X-射线荧光光谱仪(XRF),现有日本Rigaku公司生产的ZSX primus波长色散型XRF一台,及配套所必须的电源设备、冷循环水设备和前处理熔样机等。X射线荧光光谱分析技术制样简单、分析快速方便、应用广泛,可用于测定包括岩石、土壤、沉积物等在内的各种地质样品的化学组成。分析元素范围从Be(4)到U(92),最常见的是用于主量元素分析,如SiO2、Al2O3、CaO、Fe2O3T、K2O、MgO、MnO、Na2O、P2O5、TiO2、LOI等元素。 2、仪器功能和技术参数: (1) 功能:定性分析、半定量分析和定量分析; (2) X射线管:4KW超薄端窗型(30μm)、铑靶X射线管; (3) 分光晶体:LiF(200)、Ge(111)、PET、RX25、LiF(220); (4) 进样器:48位自动样品交换器; (5) 测角仪:SC:5-118度(2θ);PC:13-148度(2θ); (6) 分析元素范围:Be4-U92; (7) 线性范围:10-2 - 10-6; (8) 仪器稳定度:≤0.05%; (9) 测量误差:<5%。 3、应用和优势: XRF应用广泛,可用于岩石、矿物、土壤、植物、沉积物、冶金、矿业、钢铁、化工产品等样品中常量和痕量的定量分析。具有快速方便、制样简单、无损测量、分析元素宽、灵敏度高等优点。 X-ray Fluorescence Spectrometer (XRF) 1、I nstrument Introducation: The wavelength dispersion X-ray fluorescence spectrometer (XRF) is ZSX primus, made by Rigaku, Japan, with a set of instruments of electrical power unit, cold circulating water equipment and automatic fusion machine. XRF is widely used for geological element analysis, including rocks, soils, sediments, etc, which is simplicity and convenience of operation. Its analyzable elements range is from Be (4) to U (92). XRF is most common for the analysis of major elements, such as SiO2, Al2O3, CaO, Fe2O3T, K2O, MgO, MnO, Na2O, P2O5, TiO2 and LOI. 2、Instrument Technical Parameters: (1) Fucation: qualitative analysis, semi-quantitative analysis and quantitative analysis; (2) X-ray tube: 4KW ultrathin end-window (30μm) Rh target X-ray tube;
近红外光谱仪操作规程
NIR-Antaris II 傅立叶变换近红外光谱仪 一、工作环境 1.供电电源:AC220V±10%;50±1Hz单相交流电。 2.环境温度:15-35℃;空气相对湿度:45-80%RH。 3.仪器应置于固定的工作台上,不应有强震动源。 4.室内无电磁干扰及有害有毒气体。 二、开机 打开计算机电源开关,打开近红外光谱仪电源开关,电源指示灯(Power)亮,光谱仪开机预热1 h等仪器稳定后再使用。 三、工作流(Workflow)的建立 1.先计划好该工作流保存的路径、各样品分析报告和光谱保存路径,然后将所分析指标对应的分析模型建立到对应的文件夹中。 2.从桌面或“开始”菜单中打开RESULT-Integration软件。 3.从“文件”菜单中的“新建工作流”选项或工具栏上的“新建”工具新建一个工作流,并点击“另存为”工具将其保存到预先计划好的路径下。 4.点击工具栏上的“向导”,在弹出窗口中的“样品物质”处输入样品名称,并分别设置以下各项: ①采集 ?采集方式的选 ?背景和样品采集时的提示信息 ?采集次数、分辨率、光谱数据格式 ②测量 ?当前工作流的保存路径 ?建模方法 ?测定类型 ③报告 ?表头、表格、光谱、打印报告 ④归档
⑤点击确定,关闭当前窗口 5.在“执行”和“注释”下的文字框中输入对该工作流的描述信息,如说明该工作流的用途和方法等。 6.点击导视窗口中的各项Event前面的,将其下的子事件展开。 7.分别在导视窗口点到各项子事件,在右边的显示和参数设置窗口中设置的各项事件参数: ①设置采集项 在使用向导时已经设置过分辨率、扫描次数。在样品光谱采集时,还要看 是否使用样品杯旋转器,所以可以通过“样品规格”后面的“详细信息”按键 进入到下一个界面。对于积分球方式,如果使用,“样品杯旋转器”后面可以选 择“旋转样品杯”,不使用则选择None。 ②设置测试项 点击“详细信息”按键,选择对应的模型文件、建模所使用的方法、设置 模型测定的指标。 ③设置报告项 鼠标点击导视窗口的“报告”,点击“详细信息”按键,可设置报告的名称; 使用窗口下方的“添加”和“删除”按钮可添加或删除各项,“向上”和“向下” 按钮可对报告中的各项进行上下排序。使用右方的“新建”按钮可以新建报告 项,“详细信息”按钮可以查看和设置各项的详细参数。 ④设置存档项 在该处可设置需要存档的项目、保存路径、报告和光谱保存格式、报告和 光谱存档文件名等。 8.使用工具栏的“添加”按钮添加事件(Event)。 根据设置工作流程的需要,可以使用工具栏上的“添加”按钮添加各种Event,然后按类似于前面各步的方法设置各项参数,在导视窗口中根据需要设置好次序,以达到按既定的程序对样品进行分析的目的。 9.工作流的测试。 按前述方法建立好工作流后,可以通过工具栏上的“测试”按钮,对工作流进行测试,以检查工作流是否能够按照预定的程序运行。
红外光谱仪操作规程及注意事项
发表日期:2007年6月3日【编辑录入:admin】 1.保持室内干燥,空调和除湿机必须全天开机(保持环境条件25±10℃左右,湿度≤70%); 2.保持实验室安静和整洁,不得在实验室内进行样品化学处理,实验完毕即取出样品室内的样品。 3.经常检查干燥剂颜色,如果兰色变浅,立即更换。 4.根据样品特性以及状态,制定相应的制样方法并制样。5.测试红外光谱图时,扫描空光路背景信号和样品文件信号,经傅立叶变换得到样品红外光谱图。根据需要,打印或者保存 红外光谱图。 6.实验完毕后在记录本上记录使用情况。 7.设备停止使用时,样品室内应放置盛满干燥剂的培养皿。8.干燥剂再生:将干燥剂在烘箱内105℃烘干至兰色(约3小时)即可。 9.将压片模具、KBr晶体、液体池及其窗片放在干燥器内备用。10.液体池使用NaCl、CaF2、BaF2等晶体很脆易碎,应小心保存。11.液体池使用的KRS-5晶体剧毒,使用时避免直接接触(戴手套),打磨KRS-5晶体时避免接触或吸入KRS-5粉末,打磨的 废弃物必须妥善处理。
2010-01-12 17:11:38 来源:实验室设备信息网浏览:342次 红外光谱仪操作规程及注意事项 一、操作步骤 1.开机前准备 开机前检查实验室电源、温度和湿度等环境条件,当电压稳定,室温为21±5℃左右,湿度≤65%才能开机。 2.开机 开机时,首先打开仪器电源,稳定半小时,使得仪器能量达到最佳状态。开启电脑,并打开仪器操作平台OMNIC软件,运行Diagnostic菜单,检查仪器稳定性。 3.制样 根据样品特性以及状态,制定相应的制样方法并制样。 4.扫描和输出红外光谱图 测试红外光谱图时,先扫描空光路背景信号(Collect→Background),再扫描样品文件信号(Collect→Sample),经傅立叶变换得到样品红外光谱图。 5.关机 (1)关机时,先关闭OMNIC软件,再关闭仪器电源,最后关闭计算机并盖上仪器防尘罩。(2)在记录本记录使用情况。 二、注意事项1.测定时实验室的温度应在15~30℃,所用的电源应配备有稳压装置。2.为防止仪器受潮而影响使用寿命,红外实验室应保持干燥(相对湿度应在65%以下)。3.样品的研磨要在红外灯下进行,防止样品吸水。 4.压片用的模具用后应立即把各部分擦干净,必要时用水清洗干净并擦干,置干燥器中保存,以免锈蚀。 5.OMNI采样器使用过程中必须注意以下几点: (1)样品与Ge晶体间必须紧密接触,不留缝隙。否则红外光射到空气层就发生衰减全反
X射线荧光光谱分析法
X射线荧光光谱分析法 利用原级X射线光子或其他微观粒子激发待测物质中的原子,使之产生荧光(次级X射线)而进行物质成分分析和化学态研究的方法。在成分分析方面,X射线荧光光谱分析法是现代常规分析中的一种重要方法。 简史20世纪20年代瑞典的G.C.de赫维西和R.格洛克尔曾先后试图应用此法从事定量分析,但由于当时记录和探测仪器水平的限制,无法实现。40年代末,随着核物理探测器的改进,各种计数器相继应用在X射线的探测上,此法的实际应用才成为现实。1948年H.弗里德曼和L.S.伯克斯制成了一台波长色散的X射线荧光分析仪,此法才开始发展起来。此后,随着X射线荧光分析理论和方法的逐渐开拓和完善、仪器的自动化和计算机水平的迅速提高,60年代本法在常规分析上的重要性已充分显示出来。70年代以后,又按激发、色散和探测方法的不同,发展成为X射线光谱法(波长色散)和X 射线能谱法(能量色散)两大分支,两者的应用现已遍及各产业和科研部门。 仪器X射线荧光分析仪(见彩图)主要由激发、色散(波长和能量色散)、探测、记录和测量以及数据处理等部分组成。X射线光谱仪与X射线能谱仪两类分析仪器有其相似之处,但在色散和探测方法上却完全不同。在激发源和测量装置的要求上,两类仪器也有显著的区别。
X射线荧光分析仪按其性能和应用范围,可分为实验室用的X射线荧光光谱仪和能谱仪、小型便携式X射线荧光分析仪及工业上的专用仪器。 X射线荧光光谱仪实验室用的X射线荧光光谱仪的结构见图1 。由X射线管发射出来的原级X射线经过滤光片投射到样品上,样品随即产生荧光X射线,并和原级X射线在样品上的散射线一起,通过光阑、吸收器(可对任何波长的X射线按整数比限制进入初级准直器的X射线量)和初级准直器(索勒狭缝),然后以平行光束投射到分析晶体上。入射的荧光X射线在分析晶体上按布喇格定律衍射,衍射线和晶体的散射线一起,通过次级准直器(索勒狭缝)进入探测器,在探测器中进行光电转换,所产生的电脉冲经过放大器和脉冲幅度分析器后,即可供测量和进行数据处理用。对于不同波长的标识X射线,通过测角器以1:2的速度转动分析晶体和探测器,即可在不同的布喇格角位置上测得不同波长的X射线而作元素的定性分析。
(完整word版)Nicolet_iS5_型傅里叶变换红外光谱仪标准操作规程
本细则根据傅里叶变换红外光谱方法通则(JY?T 001-1996)和美国Nicolet公司Nicolet 380型傅里叶变换红外光谱仪操作说明书制定。 1 适用范围 本方法适用于液体、固体、气体、金属材料表面镀膜等样品。它不仅可以检测样品的分子结构特征,而且还可对混合物中各组份进行定量分析,本仪器的测量范围为4000 ~ 400cm-1。 2 术语、符号、代号 见国标(GB3100-93)。 3 方法原理 红外光谱是根据物质吸收辐射能量后引起分子振动的能级跃迁,记录跃迁过程而获得该分子的红外吸收光谱。 4 常用试剂及材料 分析纯:四氯化碳、二氯甲烷、溴化钾、氯化钠; 窗片:溴化钾、氯化钠、KRS-5(碘化铯、溴化铯合晶)。
5 检测仪器 5.1仪器技术参数 仪器名称:傅里叶变换红外光谱仪 型号:Nicolet 380 测试波数范围:4000 ~400cm-1 波数精度:≤0.1 cm-1 4cm-1分辨率就可以达到要求。 分辨率: 0.1~16cm-1,一般测试样品使用 5.2 仪器环境要求 室内温度:18℃~ 23℃ 相对湿度:≤ 50% 5.3 仪器供电需求 仪器供电电压:220V?% 交流电频率:50Hz?% 交流电零地电压:<1 V 6 检测方法 6.1 试样制备方法 6.1.1 一般注意事项 在定性分析中,所制备的样品最好使最强的吸收峰透过率为 10%左右。
Nicolet 380 型傅里叶变换红外光谱仪标准操作指导书 作者: 唐兴国审核: 丁春燕文件编号: HY-002 生效日期: 2010-11-22 最后审核日期: 2010-11-26 版次:01 修订号: 00 6.1.2 固体样品 (1)压片法:取 1~2mg的样品在玛瑙研钵中研磨成细粉末与干燥的溴化钾(A. R.级)粉末(约 100mg,粒度 200目)混合均匀,装入模具内,在压片机上压制成片测试。 玛瑙研钵压片模具 (2)溶液法:把样品溶解在适当的溶液中,注入液体池内测试。所选择的溶剂应不腐蚀池窗,在分析波数范围内没有吸收,并对溶质不产生溶剂效应。一般使用 0.1mm的液体池,溶液浓度在 10%左右为宜。 a:镜片; b:液体池部件(不含镜片); c: 装配图; d:使用方法
傅里叶红外光谱仪操作规程
傅里叶红外光谱仪操作规程 1.开机前准备 开机前检查实验室电源、温度和湿度等环境条件,当电压稳定,室温在 15~25℃、湿度≤60%才能开机。 2.开机 首先打开仪器的外置电源,稳定半小时,使得仪器能量达到最佳状态。开启电脑,并打开仪器操作平台 OMNIC软件,运行 Diagnostic菜单,检查仪器稳定性。 3.制样 根据样品特性以及状态,制定相应的制样方法并制样。固体粉末样品用 KBr 压片法制成透明的薄片;液体样品用液膜法、涂膜法或直接注入液体池内进行测定;(液膜法是在可拆液体池两片窗片之间,滴上 1-2滴液体试样,使之形成一薄的液膜;涂膜法是用刮刀取适量的试样均匀涂于 KBr窗片上,然后将另一块 窗片盖上,稍加压力,来回推移,使之形成一层均匀无气泡的液膜;沸点较低,挥发性较大的液体试样,可直接注入封闭的红外玻璃或石英液体池中,液层厚度一般为 0.01~1mm)。 4.扫描和输出红外光谱图 将制好的 KBr薄片轻轻放在锁氏样品架内,插入样品池并拉紧盖子,在软 件设置好的模式和参数下测试红外光谱图。先扫描空光路背景信号(或不放样品时的 KBr薄片,有 4个扣除空气背景的方法可供选择),再扫描样品信号,经 傅里叶变换得到样品红外光谱图。根据需要,打印或者保存红外光谱图。5.关机 (1)先关闭 OMNIC软件,再关闭仪器电源,盖上仪器防尘罩。 (2)在记录本上记录使用情况。 6.清洗压片模具和玛瑙研钵 KBr对钢制模具的平滑表面会产生极强的腐蚀性,因此模具用后应立即用水冲洗,再用去离子水冲洗三遍,用脱脂棉蘸取乙醇或丙酮擦洗各个部分,然后用电吹风吹干,保存在干燥箱内备用。玛瑙研钵的清洗与模具相同。
红外光谱仪操作规程及注意事项
红外光谱仪操作规程及注意事项 第一环境部分: 1. 保持室内干燥,空调和除湿机必须全天开机(保持环境条件不要低于20度,湿度≤65%);在南方潮湿地方,除湿机要每天都开着控制湿度,如果是由于湿度的原因,造成KBr窗片被腐蚀,是不在保修范围内的。温度变化梯度不能大于1摄氏度每小时. 2. 保持实验室清洁,不得在实验室内进行样品化学处理,实验完毕即取出样品室内的样品。 3. 一般要求红外光谱仪24小时开机,即使做不到这一点也要保证每周都开机预热三次以上,每次两个小时以上。 4.随机带的干燥剂是分子筛,可以重复使用。若仪器humidity指示灯变红色,表明干燥剂已经受潮,应倒出放到一个烧杯里在烘箱中烘干,条件是150度下连续烘24小时,降温时可置于干燥皿中以防止再度吸潮。千万不能连干燥管一起放到烘箱烘干。(由技术员负责) 5.样品室内放有盛变色硅胶的烧杯,一旦有半数以上颜色变红,必须更换硅胶。干燥剂再生:将干燥剂在烘箱内105℃烘干至兰色(约3小时)即可。 第二制样部分: 固体样品的准备 1. 样品和KBr的比例一般为1—2mg样品配上200mg的KBr。如果样品太多,测出来的吸收峰太强,如果样品太少,有些弱峰将测不出来。因不可能用天平称量,并且每种样品的对红外光的吸收程度不一致,故常凭经验取用。一般要求所测得的光谱图中绝大多数吸收峰处于10%~80%透光率范围在内。最强吸收峰的透光率如太大(如大于30%),则说明取样量太少; 相反,如最强吸收峰为接近透光率为0%,且为平头峰,则说明取样量太多,此时均应调整取样量后重新测定。一般片子厚度应在0.5mm以下,厚度大于0.5mm时,常可在光谱上观察到干涉条纹,对供试品光谱产生干扰。 2. 红外光谱测定最常用的溴化钾最好应为光学试剂级,至少也要分析纯级。溴化钾和样品用前在红外干燥箱里充分干燥,研磨3—5分钟要连同玛瑙研钵一块放到红外烘干箱里进行干燥5分钟。 3. 压片时,把样品和KBr混合物放到压片模具时,保证样品是均匀铺平在模具里,一般压力在10-15MPa, 压力太小压出来的片子不透明,压力太大容易损坏模具。一般加上压以后,保持压力1—2分钟,然后放压取片。 4、模具用后应立即把各部分用乙醇擦干净,必要时先用水清洗干净后再用乙醇擦干,置干燥器中保存,以兔锈蚀。
红外分光光度法检验标准操作规程
红外分光光度法检验标准操作规程 目的:建立红外分光光度法标准操作规程,以确保检验结果的正确性与准确性。 范围:本规程适用于红外分光光度法。 职责:检测中心、质量管理部对本规程实施负责。 内容: 1.简述 化合物受红外辐射照射后,使分子的振动和转动运动由较低能级向较高能级跃迁,从而导致对特定频率红外辐射的选择性吸收,形成特征性很强的红外吸收光谱,红外光谱又称振-转光谱。 红外光谱是鉴别物质和分析物质化学结构的有效手段,已被广泛应用于物质的定性鉴别、物相分析和定量测定,并用于研究分子间和分子内部的相互作用。 习惯上,往往把红外区分为3个区域,即近红外区(12800~4000cm,0.78~2.5m)。其中中红外区是药物分析中最常用的区域。红外吸收与物质浓度的关系在一定范围内服从于朗伯-比尔定律,因而它也是红外分光光度法定量的基础。 红外分光光度计分为色散型和傅里叶变换型两种。前者主要由光源、单色器(通常为光栅)、样品室、检测器、记录仪、控制和数据处理系统组成。以光栅为色散元件的红外分光光度计,以波数为线性刻度,以棱镜为色散元件的仪器,以波长为线性刻度。波数与波长的换算关系如下: 波数(cm-1 )= 104 /波长μm 傅里叶变换型红外光谱仪(简称FT-IR)则由光学台(包括光源、干涉仪、样品室和检测器)、记录装置和处理系统组成,由干涉图变为红外光谱需经快速傅里叶变换。该型仪器现已成为最常用的仪器。 2 红外分光光度计的检定 所用仪器应按现行国家质量与核查技术监督局“色散型红外分光光度计检定规程”、“傅里叶变换红外光谱仪检定规程”和《中国药典》附录规定,并参考仪器说明书,对仪器定期进行校正检定。
红外使用操作步骤
红外光谱仪使用操作步骤 1.把仪器主机插头和计算机主机插头插入排插中,首先打开稳压电 源的开关,然后打开排插电源;开仪器主机电源(开关在仪器后面),预热20min左右;开启计算机。 2.样品处理:(在预热的过程中操作,以便节省时间) 在玛瑙研钵中分别研磨少量KBr粉末(用来做本底),固体样品和KBr粉末的混合物(比例约1:100~300,用来测样)至2.5微米以下(大约需时2~3min),装样。取样和装样时,药品匙不能混合使用,应分别装不同的样品。测定多个样品时,中间需要清洗附件,应非常小心地拿放玛瑙研钵,附件先用自来水冲洗干净,然后用蒸馏水冲洗,再用乙醇润洗,最后放入干燥器中。 3.测试: (1)双击打开桌面omnic应用程序,当右上角Bench Status旁出现“√”,即可进行测量。 (2)测试本底: 把装本底KBr的样品槽轻轻推进光路中,点击工具栏中Collect,点击其下的collect background (或直接点击工具栏中第一个图标collect background),按ok即收集本底谱图,测试完后按yes加到窗口中。 (3)测试样品: 把装样品的样品槽轻轻推进光路中;点击工具栏中Window,点击其下的new window,新建窗口;点击工具栏中collect,点击其下
的collect sample(或直接点击工具栏中第二个图标collect sample),按ok即收集样品谱图,测试完后按yes加到窗口中。 (4)图谱处理或保存:在工具栏中File或Edit进行文件保存或编辑处理。 A.若需要做曲线平滑,点击工具栏中的Process,再点击其下的Smooth…,根据需要设定平滑的数据,平滑完后可以删掉原始的曲线,(选中原始曲线,点击工具栏中的第八个剪刀图标即可),保存平滑后的曲线。 B.若需做基线校正,应先把图转换为ABs形式(点击工具栏中第三个图标ABs即可),再进行自动基线校正(点击工具栏中第六个图标即可),校正后的图为红色,选中原有图(此时,绿色变为红色)删除,可再把图转换为透过率形式(点击工具栏中第四个图标%T即可)。 C.若想看几个图的叠加图,点击工具栏中的Window,再点击其下的New Window…,然后在这个窗口中打开想要叠加的图谱即可。 4.测试完后,关闭测试窗口,退出程序;关闭仪器主机电源,点击 “开始”,关计算机,关插排电源和稳压电源的开关,拔出插排中仪器插头。 5.清洗所用的附件,并放回原处;KBr瓶放回干燥器中。 6.使用后登记:认真记录每次开机时间、使用者、测试样品名、样 品数、机器运转情况、指导老师等。
X射线荧光光谱分析基本原理及仪器工作原理解析
X射线荧光光谱分析基本原理 当能量高于原子内层电子电子结合能的高能X射线与原子发生碰撞时,驱逐一个内层电子而出现一个空穴,使整个原子体系处于不稳定的激发态,然后自发地由能量高的状态跃迁到能量低的状态。当较外层的电子跃迁到空穴时,所释放的能量随即在原子内部被吸收而逐出较外层的另一个次级光电子,此称为俄歇效应,亦称次级光电效应或无辐射效应,所逐出的次级光电子成为俄歇电子.它的能量是具有独一特征的,与入射辐射的能量无关.当较外层的电子跃入内层空穴所释放的能量不在原子内被吸收,而是以辐射形式放出,便产生X射线荧光,其能量等于两能级之间的能量差,因此,X射线荧光的能量或波长是特征性的,与元素有一一对应的关系。如图所示: K层电子被逐出后,其空穴可以被外层中任一电子所填充,从而可产生一系列的谱线,称为K系谱线:由L层跃迁到K层辐射的X射线叫Kα射线,由M层跃迁到K层辐射的X射线叫Kβ射线……。同样,L层电子被逐出可以产生L系辐射(见图10.2)。如果入射的X射线使某元素的K层电子激发成光电子后L层电子跃迁到K层,此时就有能量ΔE释放出来,且ΔE=E K-E L,这个能量是以X射线形式释放,产生的就是Kα射线,同样还可以产生Kβ射线, L系射线等。莫斯莱(H.G.Moseley) 发现,荧光X射线的波长λ与元素的原子序数Z有关,其数学关系如下: λ=K(Z-s)-2 这就是莫斯莱定律,式中K和S是常数,因此,只要测出荧光X射线的波长,就可以知道元素的种类,这就是荧光X射线定性分析的基础。此外,荧光X射线的强度与相应元素的含量有一定的关系,据此,可以进行元素定量分析。
用X射线照射试样时,试样可以被激发出各种波长的荧光X射线,需要把混合的X射线按波长(或能量)分开,分别测量不同波长(或能量)的X射线的强度,以进行定性和定量分析,为此使用的仪器叫X 射线荧光光谱仪。由于X光具有一定波长,同时又有一定能量,因此,X射线荧光光谱仪有两种基本类型:波长色散型和能量色散型。而我们天瑞仪器公司生产的X射线荧光光谱仪就属于能量色散型的。下面是仪器的工作原理图: 能量色散型X射线荧光光谱仪工作原理 仪器工作原理 通过高压工作产生电子流打入到X光管中靶材产生初级X射线,初级X射线经过过滤和聚集射入到被测样品产生次级X射线,也就是我们通常所说的X荧光,X荧光被探测器探测到后经放大,数模转换输入到计算机,计算机计算出我们需要的结果。
Bruker红外光谱仪软件安装、使用步骤及注意事项(精)
Bruker 红外光谱仪软件安装、使用步骤及注意事项 1. OPUS软件需要工作在win2000/XP操作系统,显示器的最小分辨率要为 1024×768。 2. 需要一块 10M 或 10M/100M兼容网卡 (实际工作在 10M 模式 , 并安装好驱动程序及 TCP/IP协议。网卡的 TCP/IP协议需要指定 IP 地址 , 一般设 为 :10.10.0.100(最后一位为 2-255之间的任意整数。子网掩码为 :255.0.0.0。其它不用设置。 3. 仪器的初始 IP 地址是 10.10.0.1,可以在 IE 地址栏中键入 10.10.0.1打开仪器的硬件信息。这步操作可以检查仪器与是否联通,在日常操作过程中不会用到, 只有当仪器出现异常时,在 BRUKER 工程师的电话指导下使用。 4. 仪器使用过程中任何时候 Internet Explorer中文件菜单下的脱机工作不能被选中,否则将导致仪器不能和电脑连接。 5. windows 系统如果要安装防火墙之类软件,请在使用本仪器前关闭 , 或设置成对本仪器 IP 地址是开放的状态。 6. 在使用仪器时, IE 上所有代理服务器设置应该取消,否则仪器与电脑不能正常通讯。 7. 必须以系统管理员的身份登陆 windows 系统,才能进行 OPUS 软件的安装, 并进行用户管理设置。 8. OPUS软件安装完毕后,需要重启电脑。运行 OPUS 软件,可以点击桌面上的OPUS 图标,缺省的密码是 OPUS (大写 ,启动软件后可以更改登陆密码。 9. 每次开始测量时,点击个性化工具条的高级数据采集(或测量菜单下高级测 量选项 ,检查(或调入事先设置好的实验参数文件(例如 c:\program files\opus\xpm\MIR_TR.XPM 文件后 , 切换到检查信号页面, 应该在几秒钟后看到红色十字架形干涉图。新仪器的干涉图正常幅度(Amplitude 的绝对值应在 18000以上 (随仪器使用时间而减弱 , 位置 (position 范围应该在 58000-62000. 看见十字型干
Nicoletis红外光谱仪检定标准操作规程
1目的 建立Nicoletis5红外光谱仪的检定规程,确保仪器的性能可靠和测量的准确性。 2范围 适用于Nicoletis5红外光谱仪的检定。 3职责 质量检验部负责执行此规程,质量保证部负责监督实施。 4定义 无。 5内容 5.1检定项目和技术要求 5.2检定环境 5.2.1环境温度:16~25℃,相对湿度:≤60%; 5.2.2仪器应置于平稳的工作台上,不应有强光、强气流、强烈振动和强电磁干扰;5.2.3环境无腐蚀性气体、烟尘干扰;供电电源电压220V±22V,频率50Hz±1Hz。 5.3标准物质
聚苯乙烯膜红外波长标准物质。 5.4检定内容 5.4.1波数示值误差与波数重复性 Nicoletis5红外光谱仪扫描范围为4000cm-1~400cm-1,分辨率为4.0cm-1,常用扫描速度,扫描次数为15.待Nicoletis5红外光谱仪稳定后,采集空气本底背景,扫描聚苯乙烯红外波长标准物质,测量3027cm-1,2851cm-1,1601cm-1,1028cm-1,907cm-15个主要吸收峰。重复测量3次。按公式(1)计算,取△V绝对值最大值为波数示值误差。按公式(2)计算,取δv绝对值最大值为波数重复性。 ?v=?v i-v(1) δv=v max-v min(2) 式中:?v——波数示值误差,cm-1; δv——波数重复性,cm-1; ?vi——第i峰值波数测量平均值,cm-1; v——第i峰波数标准值,cm-1; v max——第i峰波数测量最大值,cm-1; v min——第i峰波数测量最小值,cm-1。 5.4.2透射比重复性 在T绝对值最大值为透射比重复性。 R T=T max-T min(3) 式中:R T——透射比重复性,%; T max——聚苯乙烯峰值透射比最大值,%; T min——聚苯乙烯峰值透射比最小值,%; 5.4.3分辨力 分辨苯环特征吸收峰的个数 -1~2800cm-1范围内,谱图可分辨的吸收峰的个数,见图1。 分辨深度 -1(峰)与2870cm-1(谷)之间的峰谷深度和1583cm-1(峰)和1589cm-1(谷)之间的峰谷深度,用T表示。见图2和图3。 5.4.3.3半高宽
X射线荧光光谱分析基本原理
X射线荧光光谱分析 X射线是一种电磁辐射,其波长介于紫外线和γ射线之间。它的波长没有一个严格的界限,一般来说是指波长为0.001-50nm的电磁辐射。对分析化学家来说,最感兴趣的波段是0.01-24nm,0.01nm左右是超铀元素的K系谱线,24nm则是最轻元素Li的K系谱线。1923年赫维西(Hevesy, G. Von)提出了应用X射线荧光光谱进行定量分析,但由于受到当时探测技术水平的限制,该法并未得到实际应用,直到20世纪40年代后期,随着X射线管、分光技术和半导体探测器技术的改进,X荧光分析才开始进入蓬勃发展的时期,成为一种极为重要的分析手段。 1.1 X射线荧光光谱分析的基本原理 当能量高于原子内层电子结合能的高能X射线与原子发生碰撞时,驱逐一个内层电子而出现一个空穴,使整个原子体系处于不稳定的激发态,激发态原子寿命约为10-12-10-14s,然后自发地由能量高的状态跃迁到能量低的状态。这个过程称为驰豫过程。驰豫过程既可以是非辐射跃迁,也可以是辐射跃迁。当较外层的电子跃迁到空穴时,所释放的能量随即在原子内部被吸收而逐出较外层的另一个次级光电子,此称为俄歇效应,亦称次级光电效应或无辐射效应,所逐出的次级光电子称为俄歇电子。它的能量是特征的,与入射辐射的能量无关。当较外层的电子跃入内层空穴所释放的能量不在原子内被吸收,而是以辐射形式放出,便产生X射线荧光,其能量等于两能级之间的能量差。因此,X射线荧光的能量或波长是特征性的,与元素有一一对应的关系。图1-1给出了X射线荧光和俄歇电子产生过程示意图。
K层电子被逐出后,其空穴可以被外层中任一电子所填充,从而可产生一系列的谱线,称为K系谱线:由L层跃迁到K层辐射的X射线叫Kα射线,由M层跃迁到K层辐射的X射线叫Kβ射线……。同样,L层电子被逐出可以产生L系辐射(见图1-2)。
Nicolet is5红外光谱仪检定标准操作规程
1 目的 建立Nicolet is5红外光谱仪的检定规程,确保仪器的性能可靠和测量的准确性。 2 范围 适用于Nicolet is5红外光谱仪的检定。 3 职责 质量检验部负责执行此规程,质量保证部负责监督实施。 4 定义 无。 5 内容 5.1 检定项目和技术要求 5.2 检定环境 5.2.1 环境温度:16~25℃,相对湿度:≤60%; 5.2.2 仪器应置于平稳的工作台上,不应有强光、强气流、强烈振动和强电磁干扰; 5.2.3 环境无腐蚀性气体、烟尘干扰;供电电源电压220V±22V,频率50Hz±1Hz。 5.3 标准物质 聚苯乙烯膜红外波长标准物质。
5.4 检定内容 5.4.1 波数示值误差与波数重复性 Nicolet is5红外光谱仪扫描范围为4000cm-1~400cm-1,分辨率为4.0cm-1,常用扫描速度,扫描次数为15.待Nicolet is5红外光谱仪稳定后,采集空气本底背景,扫描聚苯乙烯红外波长标准物质,测量3027cm-1,2851cm-1,1601cm-1,1028cm-1,907cm-15个主要吸收峰。重复测量3次。按公式(1)计算,取△V绝对值最大值为波数示值误差。按公式(2)计算,取δv绝对值最大值为波数重复性。 ?v=?v i-v(1) δv=v max-v min (2)式中:?v ——波数示值误差,cm-1; δv ——波数重复性,cm-1; ?vi ——第i峰值波数测量平均值,cm-1; v ——第i峰波数标准值,cm-1; v max——第i峰波数测量最大值,cm-1; v min——第i峰波数测量最小值,cm-1。 5.4.2 透射比重复性 在5.4.1取得的测量谱图中,选取峰值透射比分别为10%,20%,40%的主要吸收峰,读取峰值得透射比,按公式(3)计算,取R T绝对值最大值为透射比重复性。 R T=T max-T min (3)式中:R T ——透射比重复性,%; T max ——聚苯乙烯峰值透射比最大值,%; T min ——聚苯乙烯峰值透射比最小值,%; 5.4.3 分辨力 5.4.3.1 分辨苯环特征吸收峰的个数 在5.4.1取得的测量谱图中,检查并记录波数在3200cm-1~2800cm-1范围内,谱图可分辨的吸收峰的个数,见图1。 5.4.3.2 分辨深度 在5.4.1取得的测量谱图中,测量2851cm-1(峰)与2870cm-1(谷)之间的峰谷深度和1583cm-1(峰)和1589 cm-1(谷)之间的峰谷深度,用T表示。见图2和图3。
红外光谱仪标准操作规程
目的:建立尼高力IS5型红外光谱仪标准操作规程,规范检验人员的操作。: 范围:适用于本公司尼高力IS5型红外光谱仪的操作。 职责:质量管理部、QC. 内容: 1系统组成:本系统由主机,OMNIC光谱数据工作站和电脑等组成,另外还包括打印机、不间断电源等辅助设备。 应在120℃干燥4h,样品在105℃干燥4h,完成后放入干燥2操作前准备,KB r 器内备用。 2.1根据检验样品特性进行处理 压片法:取样品约1mg,置于玛瑙研钵中,一个方向均匀研磨,样品粒径小KB r 一个方向均匀研磨,粒径应小于2.5um。装入压片于2.5um,然后加入约100mg KB r 磨具约60mg,放入便携式压片机,进行压片,样片应平整透明。 涂膜法:将样品溶于不含水的溶剂中,如氯仿、甲醇、无水乙醇,滴加在盐片上,挥干溶剂后,进行检测。 薄膜法:将液体样品均匀涂于盐片上,然后盖上令一个盐片,稍加用力,来回推移,使之形成一层均匀无气泡的液膜,进行检测。 糊状法:将样品约1mg,置于玛瑙研钵中,一个方向均匀研磨,样品粒度小于2.5um,滴加石蜡糊或荧光湖,充分研磨,涂抹于盐片上,进行检测。 2.3 检查仪器各部件的电源线、数据线是否连接正常。 2.4准备相应的文件,如仪器操作规程、仪器使用记录、检验原始记录等。 2.5准备其它辅助用品。 3 开机:开启仪器开关,会听到“滋滋”声,蓝色指示灯闪烁,仪器需预热30min。打开电脑显示器、主机电源开关。点击电脑桌面“OMNIC”图标,软件右上角显示绿色对号,证明已联机。
4实验设置:点击上面工具栏“实验设置”图标,扫描次数为16次,选择“采集样品前采集背景”按钮,点击“确定”。 5 采集样品 5.1 点击软件上面工具栏的“采集样品”,在弹出的小窗口输入样品名称、批号,点击“确定”,先进行背景扫描。 5.2将样品取出固定于样品架,放入仪器,点击“确定”,开始扫描。 5.3 在左上角弹出窗口中点击“确定”,将谱图添加于当前窗口。 6红外谱图处理分析 6.1校正,点击“自动基线校正”,此时谱图会平整美观,点击校正前的谱图,谱图线变红色,然后点击“Ctrl+Delete”将原谱图删除,保留校正后谱图。 6.2转换,点击“数据处理”下拉菜单中的“透过率”,将谱图由吸光度转换为透过率。 6.3显示:下拉菜单“显示”选择“显示范围”,在x轴输入4000-400cm-1,在y 轴输入0-100%。 6.4标峰,点击“谱图分析”下拉菜单中的“标峰”,通过调节显示主要特征峰,然后点击“替代”,此“替代”必须执行,否则其他操作不能进行。 6.5使用谱图下方标注工具“T”,对“标峰”中未统一标出的峰进行单独标注或删除,谱图处理完成后,保存。 7谱图检索 7.1下拉菜单“谱图分析”选择“检索设置”,将“可选谱库和谱库组”全部加入“已选谱库和谱库组”,点击“确定”。 7.2点击下拉菜单“谱图分析”选择“谱图检索”,此时会看到样品谱图与谱图库进行对比。完成后可看到与标准谱图库中谱图的相似度与谱图名称。 8报告 8.1添加谱图库标准谱图,将谱图检索中标准谱图复制并粘贴于样品谱图窗口中,点击工具栏中“分层谱图显示”,窗口中分层显示标准谱图与样品谱图。 8.2点击下拉菜单“报告”选择“报告模板”,在弹出的小窗口中选择模板,并点击“编辑”,同时出现新的编辑窗口。 8.3在新窗口中输入样品名称、批号、实验仪器、型号、检测日期,注明标准谱
近红外色谱仪操作规范
GLG集团(中国)检测中心 1.目的:制定Spectrum 100N系列近红外色谱仪的使用操作规程 2.范围:本规程适用于Spectrum 100N系列近红外色谱仪的操作 3.职责:质控部检验人员对本规程负责 4.规程 4.1 Spectrum 100N系列近红外色谱仪光谱采集基本操作 4.1.1 开机操作。 4.1.1.1 打开电源,预热0.5—1小时; 4.1.1.2点击显示器桌面的图标,进入登录界面; 4.1.1.3用户名为sp100,密码为空,点击确定进入仪器连接状态的选择; 4.1.1.4 仪器连接类型选择sp100,Activate IR Assistant选择NO,点击确定进入光谱采集界面。 4.2 光谱采集 4.2.1点击工具栏上的setup进入光谱保存位置的设置,点击Options; 4.2.2点击Spectra后面的文件保存位置,然后点击Browse,手工修改光谱保存位置,按光谱采集日期将光谱图统一放在晶体检测目录下。如:今天是11月28号则光谱保存位置为D盘目录下晶体检测文件夹下的11_28的文件夹
中,点击OK; 4.2.3点击标题栏中的按钮,进入Scan and Instrument Setup 界面,将样品名字设为BJ;点击Scan,修改Options中扫描类型为background,NIR中选择lower;点击Instrument,设置分辨率为8cm1 ,点击Advanced,勾选,A VI; CO/2 H O 2 4.2.4点击标题栏按钮,点击A VI校正,点击继续,A VI校正完成后点击确定、退出,返回到Scan and Instrument Setup 界面,点击Apply、Start、Scan进行背景的扫描。 4.2.5点击标题栏中的按钮,进入Scan and Instrument Setup 界面,填写样品名字,点击Apply、Start、Scan 进行样品的扫描。 4.3 样品含量的检测 4.3.1 点击桌面上的图标,点选Existing Method ,点击确定; 4.3.2 点击STV.MD,点击确定进入样品STV含量的检测; 4.3.3点击标题栏中的按钮,点击Browse,选择光谱保存位置,选择想要预测的光谱,点击确定,记录预测的数值; 4.3.4 点击Open,选择RA.MD,点击确定进入样品RA含量的检测; 4.3.5点击标题栏中的按钮,点击Browse,选择光谱保存位置,选择想要预测的光谱,点击确定,记录预测的数值; 4.4 样品的制备
傅里叶红外光谱仪操作规程
傅立叶变换红外光谱仪 操作规程 一、主要技术指标 1、仪器型号:Nicolet 6700 2、扫描范围:4000 cm-1~ 400cm-1 3、最小精度:1cm-1 4、检测器:DTGS 5、分束器:多层镀膜溴化钾 6、光源:EverGlo光源 二、环境条件 1、电源要求: 仪器供电电压:220V±10%,频率50Hz±10% 2、温湿度要求: 室内温度18℃~25℃相对湿度≤60% 为保证仪器达到较高的控温精度,应保证稳定室温; 实验室保持抽湿状态,以维持空气干燥,且不宜开空调。 样品室窗门应轻开轻关,避免仪器振动受损。 三、试验步骤 1、标样质量浓度曲线的绘制 (1)配制系列浓度的标液:分别称取脂肪酸甲酯0.0100g、0.0200g、 0.0300g、0.0400g和0.0500g于10mL容量瓶中,加入少量环己烷摇匀, 再加环己烷至刻线位置。分别得到质量浓度为1g/L、2 g/L、3 g/L、4 g/L、 5 g/L的脂肪酸甲酯标准溶液。 (2)按以下试验操作步骤2扫描以上所配制的各个不同质量浓度的脂肪酸甲酯标准溶液,分别得到其红外谱图。 (3)打开OMNIC分析软件,将所得的不同浓度的标样图谱以及相应的浓度数值等输入该软件,绘制脂肪酸甲酯质量浓度曲线,保存。 2、试验操作: (1)开机时,首先打开仪器电源,稳定半小时,使得仪器能量达到最佳状态。 (2)开启电脑,并打开仪器操作平台OMNIC软件,运行Diagnostic 菜单,设置实验参数并检查仪器稳定性。
(3)扫描背景谱图:用环己烷反复清洗样品池(一般为3次),扫描环己烷红外谱图并保存。 (4)稀释待测试样,用稀释过的待测试样润洗样品池(2到3次)。然后向样品池中加满试样,以环己烷为背景对试样进行扫描得到其红外谱图并保存。 (5)每个样品重复进行上述(3)(4)两步骤进行平行测定。 3、试验数据分析: (1)打开OMNIC分析软件,调取试验所得的试样谱图,与标样数据对比分析,得到试样中待测物的质量浓度。 (2)试验结束后,并依次关闭OMNIC软件及仪器、主机的电源,清洗样品池,使仪器周围保持干净整洁。 四、注意事项及维护保养 1、实验室必须有良好的接地。 2、在仪器使用过程中,请经常检查仪器内部的湿度指示,Nicolet系 列用户可用软件检查干燥剂湿度是否过关。若干燥剂颜色变浅,请 及时将干燥剂在烘箱里烘干。 3、每次做完样品后,在样品仓内放一杯干燥硅胶,以保持样品仓的干 燥并同时保护两边的KBr窗片。 4、仪器长时间不使用时,间隔几天开启仪器一段时间,使仪器处于通 电状态,可防止仪器受潮。 5、每次做完试验,用布罩将仪器盖好。
- [学习]傅立叶红外光谱仪操作步骤图
- [学习]傅立叶红外光谱仪操作步骤
- 岛津IRAffinity_1S傅里叶红外光度计操作规程完整
- 红外光谱仪的基本操作
- 傅立叶红外光谱仪操作步骤_图文.ppt
- Nicoletis5红外光谱仪检定标准操作规程.docx
- 红外光谱操作规程
- Bruker红外光谱仪软件安装、使用步骤及注意事项(精)
- TENSOR27红外光谱仪操作与维护规程完整
- 红外光谱仪标准操作规程
- Nicolet_iS5_型傅里叶变换红外光谱仪标准操作规程
- 红外光谱仪操作规程及注意事项
- (优选)红外光谱仪的基本操作
- 傅里叶红外光谱仪操作规程
- 傅立叶红外光谱仪操作步骤
- 傅里叶红外光谱仪工作原理、步骤、注意事项
- 红外光谱仪操作步骤-透射
- 傅里叶红外光谱仪操作规程.pdf
- 傅立叶变换红外光谱仪的原理和应用指导书
- 傅里叶红外光谱仪操作规程(优.选)