realtimePCR和RT-PCR详解及其区别

real-time PCR技术的原理及应用
摘要:一、实时荧光定量PCR原理(一)定义:在PCR反应体系
中加入荧光基团,利用荧光信号累积实时监测整个PCR进程,最后通过标准曲线对未知模板进行定量分析的方法。(二)实时原理1、常规PCR技术:对PCR扩增反应的终点产物进行定量和定性分析无法对起始模板准
一、实时荧光定量PCR原理
(一)定义:在PCR反应体系中加入荧光基团,利用荧光信号累积实时监测整个PCR进程,最后通过标准曲线对未知模板进行定量分析的方法。
(二)实时原理
1、常规PCR技术:
对PCR扩增反应的终点产物进行定量和定性分析无法对起始模板准确定量,无法对扩增反应实时检测。
2、实时定量PCR技术:
利用荧光信号的变化实时检测PCR扩增反应中每一个循环扩增产物量的变化,通过Ct值和标准曲线的分析对起始模板进行定量分析
3、如何对起始模板定量?
通过Ct值和标准曲线对起始模板进行定量分析.
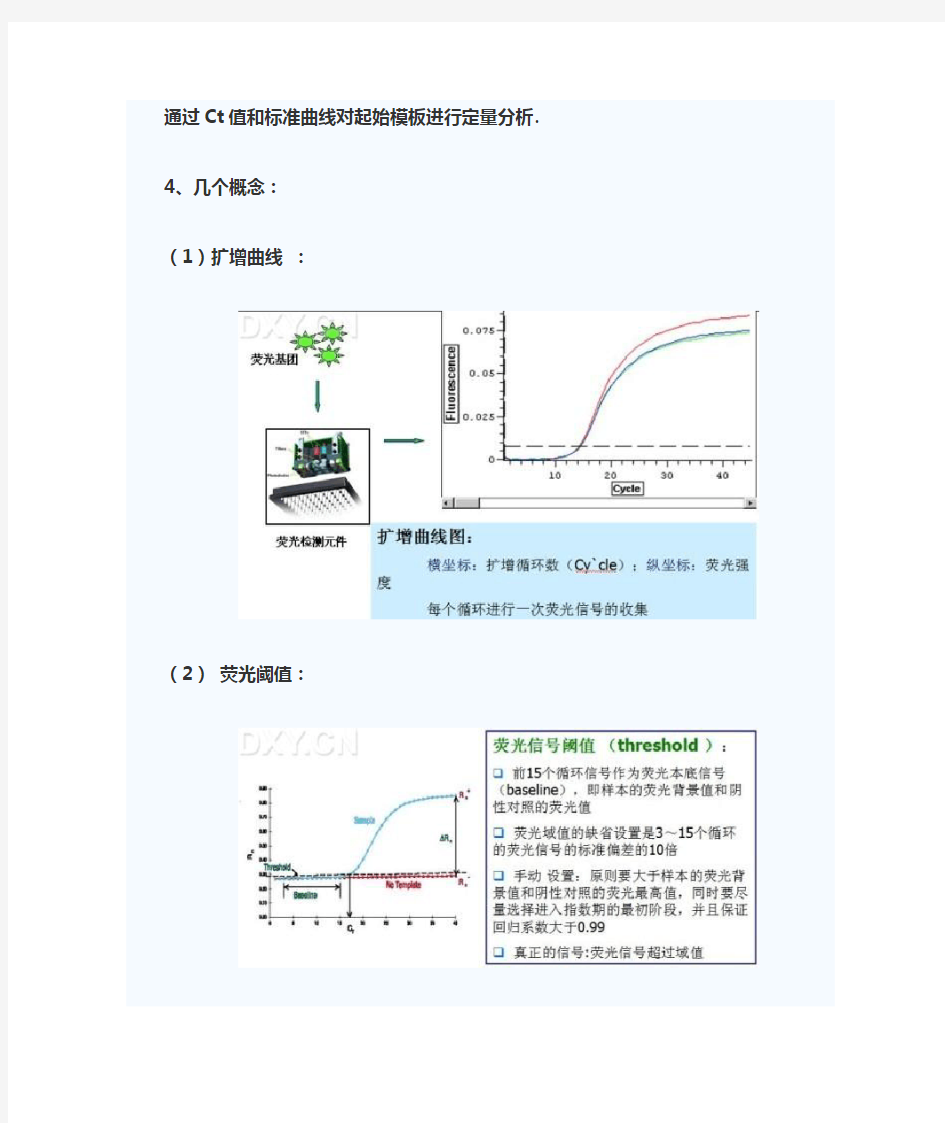
4、几个概念:
(1)扩增曲线:
(2)荧光阈值:
(3)Ct值:
CT值的重现性:
5、定量原理:
理想的PCR反应:X=X0*2n
非理想的PCR反应:X=X0 (1+Ex)n
n:扩增反应的循环次数
X:第n次循环后的产物量
X0:初始模板量
Ex:扩增效率
5、标准曲线
6、绝对定量
1)确定未知样品的C(t)值
2)通过标准曲线由未知样品的C(t)值推算出其初始量
7、DNA的荧光标记:
二、实时荧光定量PCR的几种方法介绍
方法一:SYBR Green法
(一)工作原理
1、SYBR Green 能结合到双链DNA的小沟部位
2、SYBR Green 只有和双链DNA结合后才发荧光
3、变性时,DNA双链分开,无荧光
4、复性和延伸时,形成双链DNA,SYBR Green 发荧光,在此阶段采集荧光信号。
PCR反应体系的建立及优化:
1、SYBR Green 使用浓度:太高抑制Taq酶活性,太低,荧光信号太弱,不易检测
2、Primer:引物的特异性高,否则扩增有杂带,定量不准
3、MgCl2的浓度:可以降低到1.5mM,以减少非特异性产物
4、反应Buffer 体系的优化
5、反应温度和时间参数:由酶和引物决定
6、其他与常规PCR相同
(二)应用范围
1、起始模板的测定;
2、基因型的分析;
3、融解曲线分析:可以优化PCR反应的条件,对常规PCR
有指导意义,如对primer的评价;可以区分单一引物、引物二聚体、变异产物、多种产物。
(三)优点及缺点
优点:对DNA模板没有选择性;适用于任何DNA;使用方便;不必设计复杂探针;非常灵敏;便宜。
缺点:容易与非特异性双链DNA结合,产生假阳性;但可以通过融解曲线的分析,优化反应条件;对引物特异性要求较高。
方法二:TaqMan---水解型杂交探针
**5′端标记有报告基团(Reporter, R) ,如FAM、VIC等
**3′端标记有荧光淬灭基团(Quencher, Q)
** 探针完整,R所发射的荧光能量被Q基团吸收,无荧光,
R与Q分开,发荧光
**Taq酶有5′→3′外切核酸酶活性,可水解探针
(一)工作原理
注意:每扩增一条DNA分子,释放一个荧光信号,可以在循环过程中任一点检测荧光
PCR反应的建立:
1、引物、探针的设计:
探针Tm为68-70℃,<30 bp, 5’不能有G,G可能会淬灭荧光素,
引物尽量靠近探针,扩增片段<400 bp,引物Tm为59-60℃
2、反应参数的确定:
一般为:94 ℃,10-20S
60℃,30-60S(Taq酶5′→3′外切核酸酶活性在60℃最高)
也可通过温度梯度优化退火温度72 ℃,45 S,
3、优化引物和探针浓度:获得最小Ct值,信号/背景比值的最大值
引物浓度:50-900nM
探针浓度:50-250nM
4、其他与常规PCR相同
(二)优缺点
优点:
对目标序列的高特异性
------阴性结果确定
设计相对简单
------与目标序列某一区域互补
重复性比较好
缺点:
只适合一个特定的目标;
委托公司标记,价格较高;
不易找到本底低的探针
Real-time PCR与RT-PCR比较
2009-08-20 16:50:02 来源:未知【大中小】评论: 条
摘要:Real-time PCR与RT-PCR是两种不同的PCR方法,适用范围也不同。R eal-time PCR中文译作实时聚合酶链反应,是一种最
新发展的定量PCR技术。该技术借助于荧光信号来检测PCR产物,一方面提高了灵敏度,另一方面还可以做到PCR每循环一次就收集一个数据,建立实时扩
Real-time PCR与RT-PCR是两种不同的PCR方法,适用范围也不同。
Real-time PCR中文译作“实时聚合酶链反应”,是一种最新发展的定量PCR技术。该技术借助于荧光信号来检测PCR产物,一方面提高了灵敏度,另一方面还可以做到PCR每循环一次就收集一个数据,建立实时扩增曲线,准确地确定CT值,从而根据CT值确定起始DNA拷贝数,做到了真正意义上的DNA定量,较以往常用的终点定量的方法更加准确。根据所使用的技术不同,荧光定量PCR又可以分为TaqMan探针和SYBR Green I荧光染料两种方法。
Real-time PCR所采用的专用PCR仪能够自动在每个循环的特定阶段对反应体系的荧光强度进行检测,实时的记录荧光强度的改变,从而对样品的浓度进行精确的定量。
RT-PCR是Reverse transcription PCR的简称,中文译作“逆转录聚合酶链反应”,是将RNA的反转录(RT)和cDNA的聚合酶链式扩增(PCR)相结合的技术。首先经反转录酶的作用从RNA合成cDNA,再以cDNA为模板,扩增合成目的片段。RT-PCR技术灵敏而且用途广泛,可用于检测细胞中基因表达水平,细胞中RNA病毒的含量和直接克隆特定基因的cDNA序列。作为模板的RNA可以是总RNA、mRNA或体外转录的RNA产物。无论使用何种RNA,关键是确保RNA中无RNA酶和基因组DNA的污染。用于反转录的引
物可视实验的具体情况选择随机引物、Oligo dT 及基因特异性引物中的一种。对于短的不具有发卡结构的真核细胞mRNA,三种都可。
但二者的基本原理是相同的,即PCR技术。
RT-PCR原理与实验技术
2009-08-20 16:25:05 来源:未知【大中小】评论: 条
摘要:一、知识背景:1、基因表达:DNA RNA Protein 单拷贝基因表达存在逐步放大机制,如一个蚕丝心蛋白基因104个丝心蛋白mRNA(每个mRNA存活4d,可以合成105个丝心蛋白)共合成109个丝心蛋白。因此单拷贝基因的mRNA表达水平对于其功能水平的调控是非常重要的
一、知识背景:
1、基因表达:DNA RNA Protein
单拷贝基因表达存在逐步放大机制,如一个蚕丝心蛋白基因104个丝心蛋白mRNA(每个mRNA存活4d,可以合成105个丝心蛋白)共合成109个丝心蛋白。因此单拷贝基因的mRNA表达水平对于其功能水平的调控是非常重要的。
2、PCR技术(Polymerase chain reaction):即聚合酶链式反应。
在模板、引物和四种脱氧核苷酸存在的条件下依赖于DNA聚合酶的酶促反应,其特异性由两个人工合成的引物序列决定。反应分三步:A、变性:通过加热使DNA双螺旋的氢键断裂,形成单链DNA;
B、退火:将反应混合液冷却至某一温度,使引物与模板结合。
C、延伸:在DNA聚合酶和dNTPs及Mg2+存在下,退火引物沿5‘3’方向延伸。
以上三步为一个循环,如此反复。
3、逆转录酶和RT-PCR
逆转录酶(reverse transcriptase)是存在于RNA病毒体内的依赖RNA的DNA聚合酶,至少具有以下三种活性:
1、依赖RNA的DNA聚合酶活性:以RNA为模板合成cDNA第一条链;
2、Rnase水解活性:水解RNANA杂合体中的RNA;
3、依赖DNA的DNA聚合酶活性:以第一条DNA链为模板合成互补的双链cDNA.
二、RT-PCR的准备:
1、引物的设计及其原则:
1)引物的特异性决定PCR反应特异性。因此引物设计是否合理对于整个实验有着至关重要的影响。在引物设计时要充分考虑到可能存在的同源序列,同种蛋白的不同亚型,不同的mRNA剪切方式以及可能存在的hnRNA对引物的特异性的影响。尽量选择覆盖相连两个内含子的引物,或者在目的蛋白表达过程中特异存在而在其他亚型中不存在的内含子。
2)引物设计原则的把握:引物设计原则包括
a、引物长度:一般为15~30bp ,引物太短会影响PCR的特异性,
引物太长PCR的最适延伸温度会超过Taq酶的最适温度,也影响反应的特异性。
b、碱基分布:四种碱基最好应随机分布,避免嘌呤或嘧啶的聚集存在,特别是连续出现3个以上的单一碱基。GC含量(Tm值):40%~60%,PCR扩增的复性温度一般是较低Tm值减去5~10度。
c、3‘端要求:3’端必须与模板严格互补,不能进行任何修饰,也不能有形成任何二级结构的可能。末位碱基是A时错配的引发效率最低,G、C居中间,因此引物的3’端最好选用A、G、C而尽可能避免连续出现两个以上的T。
d、引物自身二级结构:引物自身不应存在互补序列,否则会自身折叠成发夹状结构或引物自身复性。
e、引物之间的二级结构:两引物之间不应有多于4个连续碱基互补,3’端不应超过2个。
f、同源序列:引物与非特异扩增序列的同源性应小于连续8个的互补碱基存在。
g、5’端无严格限制:5’末端碱基可以游离,但最好是G或C,使PCR 产物的末端结合稳定。还可以进行特异修饰(标记、酶切位点等)等等。
根据实验目的选择适当的引物。常用引物设计软件如Primer5.0,Oligo6.0等对于这些条件都可以自行设置。
2、耗材:实验所用的接触样品的耗材如冻存管、枪头、EP管之类事先都需经过0.1%DEPC水浸泡处理,除去RNA酶,防止操作过程中
RNA降解。然后经高压灭活(灭菌和灭活DEPC)。
3、试剂准备:变性液、水饱和酚、乙酸钠、氯仿、异丙醇、75%酒精、经DEPC处理并高压的水。
三、RNA的提取方法:
RT-PCR中从细胞分离的RNA的质量至关重要,包括RNA的纯度和完整性。RNA分离的最关键因素是尽量减少RNA酶的污染。但RNA酶活性非常稳定,分布广泛,除细胞内源性RNA酶外,环境中也存在大量RNA酶。因此在提取RNA时,应尽量创造一个无RNA 酶的环境,包括去除外源性RNA酶污染和抑制内源性RNA酶活性,主要是采用焦碳酸二乙酯(DEPC)去除外源性RNA酶,通过RNA 酶的阻抑蛋白Rnasin和强力的蛋白质变性剂如异硫氰酸胍抑制内源性RNA酶。
RT-PCR步骤:
1、RNA的提取:
2、cDNA的合成:
逆转录体系的组成:
DEPC处理水8ul
10mM dNTPs 2.5ul
Random primers 0.4ul
RNasin 0.5ul
总RNA 2.5-3ug
70℃5min
速置冰水中冷却
再加:5*逆转录buffer 5ul
逆转录酶(M-MLV) 1ul(200U)
加水至25ul
37 ℃60min
90℃5min
3、PCR扩增:
50ul PCR体系的组成:
10×PCRbuffer 5ul
MgCl2 3ul(2.0mM)
10mMdNTPs 1ul
sense primer 1ul(1umol/l)
antisense primer 1ul(1umol/l)
cDNA 1.5ul
DEPC处理水36.7ul
Taq 酶0.8ul(2.4U)
总体积50ul
4、PCR反应条件:
94 ℃5min
94 ℃1min
退火温度40sec
72 ℃50sec 2 29 cycles
72 ℃7min
4 ℃0sec
四、PCR条件的优化:
PCR条件的优化主要是①引物退火温度的调节,一般在引物设计软件推荐温度的上下2 ℃变化寻找最佳退火温度。②此外Mg2+浓度的调节也非常重要,通常最佳浓度为2mM左右。③引物和模板的量等。
五、产物的电泳和结果的测定:
根据产物长度制作适宜浓度的琼脂糖凝胶(不同长度产物所需凝胶浓度)
取适量PCR产物,加上样缓冲液充分混合,点样于事先做好的琼脂糖凝胶点样孔中,10V/cm电泳45-60min。成像系统成像分析。分离不同大小DNA片段的合适琼脂糖浓度
琼脂糖浓度(%)线性DNA片段的有效分离范围(kb)0.5 1-30
0.7 0.8-12
1.0 0.5-10
1.2 0.4-7
1.5 0.2-3
六、需注意的问题:
1、注意避免有毒、有害试剂伤害自己和他人:氯仿、溴化乙锭(EB)、酚、异硫氰酸胍、紫外线等
2、注意避免试剂污染
3、始终注意避免RNA酶的污染:
4、保管好自己专用的试剂,公用试剂保管好,相互协调,注意实验室卫生
Real time PCR 跟RT-PCR有什么区别
关键词:real time PCR RT-PCR区别?2008-07-23 00:00 来源:丁香园点击次数:4932
实时荧光定量PCR原理
所谓实时荧光定量PCR技术,是指在PCR反应体系中加入荧光基团,利用荧光信号积累实时监测整个PCR进程,最后通过标准曲线对未知模板进行定量分析的方法。
1.内标在传统定量中的作用
由于传统定量方法都是终点检测,即PCR到达平台期后进行检测,而PCR经过对数期扩增到达平台期时,检测重现性极差。同一个模
板在96孔PCR仪上做96次重复实验,所得结果有很大差异,因此无法直接从终点产物量推算出起始模板量。加入内标后,可部分消除终产物定量所造成的不准确性。但即使如此,传统的定量方法也都只能算作半定量、粗略定量的方法。
2.实时荧光定量PCR无需内标
实时荧光定量PCR技术有效地解决了传统定量只能终点检测的局限,实现了每一轮循环均检测一次荧光信号的强度,并记录在电脑软件之中,通过对每个样品Ct值的计算,根据标准曲线获得定量结果。因此,实时荧光定量PCR无需内标是建立在两个基础之上的:
1)Ct值的重现性
PCR循环在到达Ct值所在的循环数时,刚刚进入真正的指数扩增期(对数期),此时微小误差尚未放大,因此Ct值的重现性极好,即同一模板不同时间扩增或同一时间不同管内扩增,得到的Ct值是恒定的。
2)Ct值与起始模板的线性关系
由于Ct值与起始模板的对数存在线性关系,可利用标准曲线对未知样品进行定量测定,因此,实时荧光定量PCR是一种采用外标准曲线定量的方法。
rtpcr(rt pcr)
rt-pcr(rt - pcr) I. Experimental apparatus and materials: 1, transfer guns: 1ml, 200 L, 20 L, 10 L, 2 L 2, suction head: 1ml, 200 L, 20 L 3, homogenate tube: 5ml 4, suction head table: put 1ml suction head one, put 20 l suction head one 5, EP tubes: 1.5ml, 0.2ml, 100 L 6, reagent bottle: 2 60ml Brown reagent bottle (wide mouth, with cover) 1 125ml white reagent bottles (with absolute ethanol) 7, 50ml, 250ml, 500ml: a 8, capacity bottle: 250ml, 500ml, 1000ml 9, test tube rack: 5ml, 1.5ml, 20 L 10, salt water bottles: 250ml, 500ml each 2 spare, one with absolute ethanol, another with DEPC water 11, aluminum lunch box: 4 12, plastic small lunch box: 1
13, large porcelain cylinder: 2 14, tin paper: a roll 15 rolls of paper: 2 rolls 16, triangle flask: with a cap, slightly larger Two, the processing and preparation of experimental equipment 1, plastic products: (including gun head, EP tube, homogenate tube, etc.) The DEPC of water from the flask into the ceramic cylinder, the plastic products by soaking them, which requires a small tip Straw DEPC into the water, and then dried overnight, high pressure, spare, before the experiment will first put the gun suction head, and a high pressure (EP tube) 2, glass products: acid soaking overnight, washed clean, dry spare foil Mongolia (DEPC blister) (wash after the first bubble 1 per thousand DEPC overnight, and then dried) 3, homogenizer: (including scissors, tweezers) first wash, and then high pressure (do not need to bubble DEPC) Three. Reagent preparation: 1, DEPC water: suck 1ml, placed in 1000ml double steamed water, with 1 per thousand DEPC water, placed in the 1000ml capacity
三种大屏技术对比
液晶拼接与背投(DLP)、等离子(PDP)的技术对比 背投原理简析 背投的实现原理很简单,在设备内部设置一部投影机,发出的图像经透镜放大后投射到屏幕背面,就是背投。正是基于这种原理诞生的背投,由于采用不同的投影机种类,主要可分为CRT(阴极射线管)、LCD(液晶)、DLP(数字光处理)等几种。CRT背投属于背投阵营中的低端产品,而其它几种背投则对应地为高端产品,其中以DLP背投最为出色,其图像清晰度、亮度、色彩、可视角度以及体积来看,均比传统CRT背投有了很大提高。以下文中所述背投均指DLP背投。
优点:廉价的低端显示方案。 缺点:体积与重量过大,长时间不间断工作,加快背光灯老化。 等离子原理简析 PDP是一种利用气体放电的显示技术,其工作原理与日光灯很相似。它采用了等离子管作为发光元件,屏幕上每一个等离子管对应一个像素,屏幕以玻璃作为基板,基板间隔一定距离,形成一个个放电空间。放电空间内充入氖、氙等混合惰性气体作为工作媒质,在两块玻璃基板的内侧面上涂有金属氧化物导电薄膜作激励电极。当向电极上加入电压,放电空间内的混合气体便发生等离子体放电现象,也称电浆效应。等离子体放电产生紫外线,紫外线激发涂有红绿蓝荧光粉的荧光屏,荧光屏发射出可见光,显现出图像。优点:颜色鲜艳、高亮度、高对比度缺点:耗电与发热量很大,严重灼伤现象,画质随时间递减。 液晶原理简析 液晶是利用液状晶体在电压的作用下发生偏转的原理。由于组成屏幕的液状晶体在同一点上可以显示红、绿、蓝三基色,或者说液晶的一个点是由三个点叠加起来的,它们按照一定的顺序排列,通过电压来刺激这些液状晶体,就可以呈现出不同的颜色,不同比例的搭配可以呈现千变万化的色彩。液晶本身是不发光的,它靠背光管来发光,因此液晶屏的取决于背光管。由于液晶采用点成像的原因,因此屏幕里面构成的点越多,成像效果越精细,纵横的点数就构成了液晶电视的分辨率,分辨率越高,效果越好。 优点:高分辨率、厚度薄、重量轻、低能耗、长寿命、无辐射。 缺点:拼接缝稍大。 液晶和等离子显示技术PK 目前主流的平板显示技术主要有液晶显示技术和PDP等离子显示技术。下面,我们就从几个方面比较一下这两种显示技术。 1.使用寿命 大屏幕显示器由于其不菲的造价,所以使用寿命成为其首要问题,理论上讲液晶和等离子显示屏的寿命都可以达到6万小时,不过由于这两种显示技术的发光原理不同,使得实际应用中差异很大。等离子显示器中的每个像素单元实际上是一个微型灯泡,由于使用白炽灯泡,图像质量会随着使用时间增长而变差,虽然目前的技术能够目前的技术能够使等离子显示器工作时间达到60,000个小时,但可能使用到20,000小时的时候背光就会出故障,导致显示质量下降一半。并且等离子如果长期播放一个固定的图像,会在屏幕上留下一个浅浅的痕迹(残影)也就是“烧屏”,例如,如果观看一信号太久,屏幕一角的台标就可能烙印在屏幕上,在观赏其它信号时仍看得到其残影。通常情况下,连续观看10~20小时就能造成看得见的残影,截至目前这个问题还没有完美的解决方法。由于液晶电视工作原理不同(利用液状晶体在电压的作用下发光成像的原理。组成屏幕的液状晶体有三种:红、绿、蓝三基色,
荧光标记mRNA差异显示技术
荧光标记mRNA差异显示技术 mRNA差异显示技术(differential display,DD)是用于研究基因的差异表达的新方法。该技术自1992年被首次报道后,即以其不可替代的优势被广泛应用于生物医学领域。在应用过程中不断得到改进,并产生了诸多衍生技术如RPA(RNA finger printing by arbitrarily primed PCR)、GDD (genomic DD)等。本文简要介绍在本试验室荧光标记差异显示技术(fluorescent DD,FDD)的应用及体会。 1.材料与方法 1.1 标本 人单核细胞系U937,细胞密度2×108/L,分对照组(N)、处理Ⅰ组(T1)、处理Ⅱ组(T 2),N用1640培养基及10%胎牛血清培养,T1用IF N-γ104U/L LPS 1μg/L、T2用IFN- γ10 U/L LPS l0μg/L分别刺激7h. 1.2 主要试剂与仪器 TRIzol试剂(GIBCO BRL)、Fluoro DD试剂盒(Genomyx)、Supersript Ⅱ逆转录酶(GIBCO BRL)、Ampli Taq DNA聚合酶(GIBCO BRL)、RNase-free DNaseI(Promega);Genomyx LR 、 Genomyx SC、DNAT hermal Cycler(Perkin Elmer) 1.3 总RNA的制备 按试剂盒提供的方法分别提取三种细胞的总RNA,以RNase-free DNase I(终浓度80 000 U/L)除去其中污染的 DNA,经甲醛变性凝胶电冰鉴定其完整性,并以紫外分光光度计检测其纯度 1.4 mRNA差异显示 1.4.1 逆转录反应 选择锚定引物[T7(dT12)AP(anchored prime rs,AP)],序列为5'ACGACTCACT ATAGGGCTTTTTTTTTTTTMN3',其中M=A/G/C。N=A /G/C/T],以总RNA为模板进行逆转录反应,每管反应体系如下:总RNA l.0μg,AP 4 pmol ,70℃5 min,加入50 mmol/L Tris-HCl(pH8.3),75 mmo l/L KCl,3 mmol/L MgCl2,10mmol /L DTT,25μmol/L,dNT Pmix(1∶1∶1∶1),SuperScriptⅡ 60 Units,总反应体系20 μl ,42℃5 min,5 0℃50 min,70℃15 min。 1.4.2 荧光标记差异显示P CR 选取与逆转录引物序列相同的带荧光物质标记的锚定引物[TMR-T7(dT12)AP],随机引物(5'ACAATTTCACACAGGAACGCT AGTT G 3'),以逆转录产物为模板,进行PCR反应。反应体系包括:20 mmol/L Tris-HCl(pH8.4),50 mmol/L KCl,3.75 mmol/L MgCl2,逆转录产物3.0 μl,50 μ mol/L dNT P mi x(1∶1∶1),0.35 μmo l/L 5'-随机引物,0.35 μmol/L 3-锚定引物,Ampli Taq 0.5 U nits,总反应体系10 μl。反应步骤如下:95℃2 min;94℃15 s,50℃ 30 s,72℃2 min,4个循环; 94℃15 s,60℃30 s,72℃ 2 min,个循环;72℃延伸7 min。 1.4.3 分离差异显示片段 配制5.6%变性聚丙烯酰胺凝胶,胶厚0.2 5 mm,大小61×33 cm。将PCR产物加4.0μl上样缓冲液,95℃变性后上样。3000V、100W 、55℃电泳4.5 h。干胶后置于Genomyx SC扫描,用系统所带的AcquireSC program软件分析处理扫描结果。 1.4.4 回收差异条带
mRNA差别显示技术
mRNA差别显示技术(DDRT-PCR) 随着PCR技术的发展,人们在此基础上建立起了一系列基于基因分离的新技术新方法。如mRNA差别显示技术(DDRT-PCR)、以及进一步改进的代表性差示分析(RAD),抑制性扣除杂交(SSH)和交互扣除RNA差别显示技术(RSDD)。 差别显示PCR是根据绝大多数真核细胞mRNA3’端具有的多聚腺苷酸尾(polyA)结构,因此可用含oligo(dT)的寡聚核苷酸为引物将不同的mRNA反转录成cDNA。该方法的创始人Liang P和Pardee A根据Poly A序列起点前2个碱基除AA外只有12种可能性的特征,设计合成了12种下游引物,称3′-锚定引物,其通式为5’-T11MN;同时为扩增出polyA 上游500bp以内所有可能性的mRNA序列,在5′端又设计了20种10bp长的随机引物。这样构成的引物对进行PCR扩增能产生出20000条左右的DNA条带,其中每一条都代表一种特定mRNA种,这一数字大体涵盖了在一定发育阶段某种细胞类型中所表达的全部mRNA。 差别显示技术自1992年建立后,一直在不断进行着改进。如在1994年,Ito等对3′端锚定引物的设计由固定两个碱基变为一个碱基固定的引物,这就使原来12种引物减至3种即可(5′T12G,5′T12A,5′T12C),这样做减少了每个mRNA样品对逆转录反应种类的需要,并且把由于简并性引起的某些RNA的代表性差和RNA数量过多现象降低到最低程度;在随后的两年中,研究人员有在3′端引物和5′随机引物末端分别加上了限制性内切酶识别位点(如Hind III酶切位点),使得5′端引物条数改为8条,长度为13bp,而3′端引物则由18个碱基组成。这样形成的24种引物对,经计算机同源性分析表明同样能覆盖全部mRNA,使实验简化,同时由于引物变长,使cDNA扩增更为有效。有的实验室采用把Bakman 公司的kit,其引物5′端为26bp,3′端为31bp,并加上了T3和T7两端的测序引物,由仪器切割差异条带后,再次扩增,并通过荧光标记,用计算机统计出结果。此外,许多学者针对该技术在****作中假阳性高等问题,还从设计对照、提取胞质RNA、更换放射性标记物以及改变PCR反应条件等等方面提出了相应解决方案,以求这一技术得到进一步完善。 DD-PCR与示差筛选、扣除杂交相比,具有很多优点: 1)速度快,较易操作; 2)由于PCR扩增技术的应用,使得低丰度mRNA的鉴定成为可能,且灵敏度高; 3)可同时比较两种以上不同来源的mRNA样品间基因表达的差异。 尽管差别显示技术有以上诸多方面的优点,但在实际操作中仍存在一些问题,主要表现在: 1)出现差别条带太多,假阳性率高达70%左右,重复性差,且对高拷贝数的mRNA具很强的倾向性; 2)扩增条带分子长度较短,一般在110~450bp之间。
显示技术对比(LCD与DLP)
LCD、DLP大屏幕显示系统技术对比 1.当前市场主流投影大屏幕显示技术比较 1.1LCD技术 液晶式投影机全称为液晶显示式(Liquid Crystal Display,缩写为LCD)投影机。一个LCD扮演一个光阀的角色,它最好能被理解为一个能够调制和控制通过面板可以发射的偏振光的总量的机构。LCDs的改进已倾向于增加透射率(光输出),但是LCD仍然局限于模拟结构。非晶硅和多晶硅是薄膜晶体管(TFT)LCDs,它需要一个晶体管来控制LCD板上的每一个象素。通过晶体管提供给LCD象素的一个电子信号改变了象素的极性。通过改变极性,通过每个象素的光的总量可以被控制来产生一个图像。 三个闭合分隔的红、绿和蓝LCD次级象素。光可以表示为垂直和水平分量,如果光定位在一个垂直取向的偏振镜上,这个偏振片扮作一个滤光片,并且只允许垂直光通过。这个系统的另一面放置了另外一个偏振片,因而光只能在水平方向通过。在路径上没有液晶时第一个偏振片将阻挡水平光而通过垂直光。当垂直光打倒第二个偏振片时,它也将被阻挡(因为第二个偏振片仅通过水平光)。这一结果是光的完全封闭状态,产生一个黑象素。当一个液晶“夹心”在两个偏振片之间时,它扮作一个偏振光的调制器或“绞扭器”。通过把一个电压加到液晶上,光的极性可以被改变,允许各种不同水平的光通过系统,基于LCD技术的投影系统使用一个单独的LCD板或者三个LCD板,一个板一种基本的颜色——红、绿和蓝。在显示在这儿的单板构造图中,小的,封闭间隔的红、绿和蓝次级象素组成一个象素。 1.2DLP技术 DLP是Digital Light Processing的英文缩写,意为数字光学处理,是一种基于美国德州仪器公司(Texas Instrumens)开发的数字微反射镜器件DMD(Digital Micromirror Device)技术的数字光学成像技术。 DLP是投影和显示信息领域的一个革命性的新方法,由数字电路驱动,是完成显示数字可视信息的最终环节。 对于影视投影显示、计算机幻灯展示或全球范围内多人通过交互技术进行合作等方面,DLP是现在和未来在数字可视通信方面的唯一选择。正如CD在音视频领域的革命一样,DLP必将带来一场视频投影领域的革命。
mRNA差别显示技术
mRNA差别显示技术 mRNA差别显示技术也称为差示反转录PCR(Differential Display of reverse Transcriptional PCR)简称为ddRT-PCR。标本检测,你提供标本,我免费给你做实验,成果归你享有Q 往圣科技3452125268 它是将mRNA反转录技术与PCR技术二者相互结合发展起来的一种RNA指纹图谱技术。目前已广泛应用于分离鉴定组织特异性表达的基因。差别基因表达(differential gene express)是细胞分化的基础。基金论文,你提供实验材料,我免费给你做实验,成果归你享有Q往圣科技3452125268 一、mRNA差别显示技术正是对组织特异性表达的基因进行分离的一种快速而行之有效的方法。该方法的基本原理是首先选取不同的组织样品或不同发育阶段的同一组织样品或同一组织样品经不同药物处理诱导的样品,经总RNA提取后,Q往圣科技3452125268提供12万种CRISPR/cas9免费送 进行mRNA反转录合成cDNA。此cDNA的合成是采用Oligo(dT)12 MN为引物,其中M为A,C,G中的任意一种,N为A,C,G或T中的任意一种,所以共有12种oligo(dT)12 MN 引物,其中M称为锚定碱基,起增大引物Tm值的作用,N称为分类碱基,对反转录进行分类。Q往圣科技3452125268提供张峰Science的CRISPR/cas9免费送 用这12种引物分别对同一总RNA样品进行cDNA合成,即进行12次不同的反转录反应,从而使反转录的cDNA具有12种类型,也就是对cDNA进行12种归类(目前较为流行的方法是进行4种归类,即M为简并碱基的形式存在),在此基础上对每一类cDNA进行随机引物和反转录引物PCR扩增,通过对组织样品的同一类cDNA的PCR选择性扩增产物凝胶电泳分析,从而反映出不同样品间基因的时间和空间上组织特异性的表达。如此众多种类的mRNA反转录产物经PCR选择扩增后其类型依然为数众多,很难用电泳系统加以快速准确地分离Q往圣科技3452125268提供Science的CRISPR/cas9免费基因慢病毒包装。因而需要对反转录产物cDNA进行归类处理,减轻不同PCR产物电泳分离的难度,提高分离的准确率。(见示意图) 实验流程:Q往圣科技3452125268提供Science的CRISPR/cas9免费质粒加慢病毒包装
rtpcr注意事项
RT-PCR技术 RT-PCR简介 RT-PCR的指数扩增是一种很灵敏的技术,可以检测很低拷贝数的RNA。 RT-PCR广泛应用于遗传病的诊断,并且可以用于定量监测某种RNA的含量。(检测基因表达的方法,参见Northern Blot法。) RT-PCR有时候也会指代实时PCR(real-time PCR)。为了与逆转录PCR 相区别,通常被写作“定量PCR”(quantitative PCR)或者 RTQ-PCR(real-time quantitative PCR)。 实时PCR 实时PCR(real-time PCR),属于定量PCR(Q-PCR)的一种,以一定时间内DNA的增幅量为基础进行DNA的定量分析。 real time PCR 的定量使用萤光色素,目前有二种方法。一种是在ds DNA (双链DNA)中插入特异的萤光色素;另一种使用一种能与增幅DNA序列中特定寡核酸序列相结合的一种萤光探针(probe)。 real time PCR 与reverse transcription PCR(反转录PCR) 相结合,能用微量的RNA来找出特定 时间、细胞、组织内的特别表达的遗传基因。这两种RT PCR的组合又被称之为“定量RT-PCR(quantitative RT-PCR)” RT-PCR技术相关试剂 oligo: 多聚体,相当于mRNA引物 AMV(M-MLV):逆转录酶 dNTP:脱氧核苷酸 RNase:RNA酶抑制剂
PCR Buffer:RT-PCR缓冲液 MgCl2:2价镁离子 (一)预变性: 破坏DNA中可能存在的较难破坏的二级结构。使DNA充分变性,减少DNA 复杂结构对扩增的影响,以利于引物更好的和模板结合,特别是对于基因组来源的DNA模板,最好不要吝啬这个步骤。此外,在一些使用热启动Taq酶的反应中,还可激活Taq酶,从而使PCR反应得以顺利进行。 (二)变性--退火--延伸循环: ①模板DNA的变性:模板DNA经加热至93℃左右一定时间后,使模板DNA双链或经PCR扩增形成的双链DNA解离,使之成为单链,以便它与引物结合,为下轮反应作准备; ②模板DNA与引物的退火(复性):模板DNA经加热变性成单链后,温度降至55℃左右,引物与模板DNA单链的互补序列配对结合; ③引物的延伸:DNA模板--引物结合物在TaqDNA聚合酶的作用下,以dNTP为 反应原料,靶序列为模板,按碱基配对与半保留复制原理,合成一条新的与模板DNA 链互补的半保留复制链。 (三)PCR仪扩增循环后72度延伸10分钟 用PCR仪扩增时,(变性.退火,延伸)循环完成后,继续72度延伸了10分钟的原因: 1.延伸时间取决于待扩增DNA片段的长度。(当然是在反应体系一定的条件下)例如,使用taqDNA聚合酶,72度时的碱基掺入率为35-100bp/s,因此延伸速率为1kb/min。 2.根据延伸速率推得,扩增1kb以内的dna片段1min即可,而3-4kb 则需要3-4min,依次照推。通常在最后一轮要适当的将延伸时间延长至 4-10min,这样做是使pcr反应完全以提高扩增产量。 3.继续72度延伸了10分钟除了可以使pcr反应完全以提高扩增产量外,还有一个作用是:在用普通taq酶进行PCR扩增时在产物末端加A尾的作用,可以直接用于TA克隆的进行。 RT-PCR的注意事项
四种新型高清显示技术对比
四种新型高清显示技术对比
四种新型高清显示技术对比 以LCD、PDP、DLP、LCoS为代表的新兴显示技术,代表了数字电视时代电视机技术发展的方向,注定成为显像管电视机的终结者。数字电视,特别是高清晰度电视机,也注定成为世界电视发展的潮流。随着我国经济水平的发展,特别是迎合2008年北京奥运会的契机,HDTV节目出现在我们身边的时间并不遥远…… 以LCD、PDP、DLP、LCoS为代表的新兴显示技术,代表了数宁电视时代电视机技术发展的方向,注定成为显像管电视机的终结者。数字电视,特别是高清晰度电视机,也注定成为世界电视发展的潮流。随着我国经济水平的发展,特别是迎合2008年北京奥运会的契机,HDTV节目出现在我们身边的时间并不遥远。下面将分别介绍LCD、PDP、DLP、LcoS 4种新兴的显示技术的优缺点和前景。 LCD——液晶电视 液晶电视和传统的显像管电视机比,液晶电视机具有很多优势:1、显示质量高,无闪烁;2、无电磁辐射;3、画面效果好,无变形,是真正的纯平显示;4、屏幕大小可伸缩性好。目前最大的LCD显示屏可以大到65英寸,小的却可以使用到数码相机和手机上。其体积和重量均比CRT要小许多。5、清晰度高,可真正实现HDTV的效果;6、数字式工作方式,更完美的表现数字图像信号;7、功耗小,只有同面积CRT电视机的1/10~1/7。 相对于同样是平板电视成员的PDP电视,LCD电视也有一些PDP电视所没有的优点: l、使用寿命更长,PDP显示器的标称寿命大多在2.5万~3万小时,而且是不可恢复的,这与LCD 显示器的5万~7.5万小时(可以通过更换背光管恢复)相比要逊色很多; 2、比PDP彩电功耗更低,更省电; 3、液晶电视作为3C产业融合的重要产品,吸引了众多IT厂商和家电厂商的共同参与,将有助于液晶电视成本的降低,将市场做大。 PDP——等离子电视 在平板电视家族中,除了LCD以外,就是PDP了。PDP,即等离子显示器,是继LCD之后的最新显示技术之一。 PDP属于“自发光”的平面显示技术.核心原理和日光灯发光原理类似,是在真空玻璃(即放电空间)中注入惰性气体,然后再利用施加电压的方式,使管内的气体产生放电,应用离子效应而释放出紫外线,照射涂覆在玻璃管管壁上的荧光粉,荧光粉就会被激发出可见光,而不同的荧光粉会被激发不同颜色的可见光。
mRNA差异显示技术
mRNA差异显示技术 mRNA differetial display 1.概述 mRNA差异显示技术(mRNA differetial display)是一种快速有效的克隆差异性表达基因的方法。 ?方法建立:1992年Liang P和Pardee首次应用DD技术对比人类乳腺癌细胞与正常细胞所表达的mRNA,以此来克隆癌细胞所特有的基因 ?目前已应用于个各领域: –农业、植物 –动物 –医学 ?胚胎发育 ?遗传病 ?药物 ?肿瘤 2.mRNA差异显示原理 ?是分离差异表达基因的一个重要方法。人类大约含有三万个不同的基因。在不同的单个细胞中只有10%的基因处于表达状态。 ?生物体在不同的生理,病理状态下,基因的表达水平不同。 ?生命过程中选择性表达的基因主要有:发育与分化、体内平衡、对攻击的应答、细胞周期调节、老化和程序性细胞死亡等。 ?差异显示获得的只是某个基因的片段,而不是直接获得全长cDNA克隆。 3.基本程序 –选择表型特性差异显著对象 –展示mRNA的差异 –基因差异 –克隆与表型差异高度相关的新基因 4.技术 ?基本技术 –逆转录(RT ) –聚合酶链反应(PCR) –凝胶电泳 5.RT-PCR ?锚定引物T12-MN,含有12个T可以结合于mRNA 3`端poly(A)尾,M为A、C、G 3种碱基之一,N为A、T、C、G 4种碱基之一。 ?荧光锚定引物,引物5`端加入T7 RNA多聚酶启动子并带有荧光分子 ----------- AAAAAAAAAA –3` mRNA ------------XAAAAAAAAAAA–3` 1/3 mRNA MTTTTTTTTTTTT(12T)Prime
RTPCR原理和实验步骤
RT—PCR原理与实验步骤 一、知识背景: 1、基因表达:DNA RNA Protein 单拷贝基因表达存在逐步放大机制,如一个蚕丝心蛋白基因 104个丝心蛋白mRNA(每个mRNA存活4d,可以合成105个丝心蛋白) 共合成109个丝心蛋白。因此单拷贝基因的mRNA表达水平对于其功能水平的调控是非常重要的。 2、PCR技术(Polymerase chain reaction):即聚合酶链式反应。 在模板、引物和四种脱氧核苷酸存在的条件下依赖于DNA聚合酶的酶促反应,其特异性由两个人工合成的引物序列决定。反应分三步: A。变性:通过加热使DNA双螺旋的氢键断裂,形成单链DNA; B.退火:将反应混合液冷却至某一温度,使引物与模板结合. C。延伸:在DNA聚合酶和dNTPs及Mg2+存在下,退火引物沿5’3’方向延伸。以上三步为一个循环,如此反复。 3、逆转录酶和RT-PCR 逆转录酶(reverse transcriptase)是存在于RNA病毒体内的依赖RNA的DNA聚合酶,至少具有以下三种活性: 1、依赖RNA的DNA聚合酶活性:以RNA为模板合成cDNA第一条链; 2、Rnase水解活性:水解RNA:DNA杂合体中的RNA; 3、依赖DNA的DNA聚合酶活性:以第一条DNA链为模板合成互补的双链cDNA。 二、RT—PCR的准备: 1。引物的设计及其原则: 1)引物的特异性决定PCR反应特异性.因此引物设计是否合理对于整个实验有着至关重要的影响。在引物设计时要充分考虑到可能存在的同源序列,同种蛋白的不同亚型,不同的mRNA剪切方式以及可能存在的hnRNA对引物的特异性的影响。尽量选择覆盖相连两个内含子的引物,或者在目的蛋白表达过程中特异存在而在其他亚型中不存在的内含子。 2) 引物设计原则的把握 引物设计原则包括: a、引物长度:一般为15~30bp ,引物太短会影响PCR的特异性,引物太长PCR的最适延伸温度会超过Taq酶的最适温度,也影响反应的特异性。 b、碱基分布:四种碱基最好应随机分布,避免嘌呤或嘧啶的聚集存在,特别是连续出现3个以上的单一碱基。GC含量(Tm值):40%~60%,PCR扩增的复性温度一般是较低Tm值减去5~10度.
差异显示 RT-PCR技术
差异显示RT-PCR技术 杨一文 (宁波诺丁汉大学,宁波315100) 摘要:差异显示RT-PCR技术是在基因转录水平上研究差异表达和性状差异的有效方法之一,该方法在生物的发育、性状和对各种生物、理化因子作用时应答过程基因表达的研究中应用十分广泛,本文对差异显示RT-PCR技术的基本原理、技术进行介绍并举例了此技术在球虫抗药性研究中的应用。 关键词:差异显示RT-PCR;球虫;抗药性 人类基因组计划证明,不同人种之间的基因有99.99%是相同的,甚至低于同一种族的不同个体的基因差异。造成这种现象的原因即是基因的选择表达,它决定着个人从发育分化,内环境稳定,细胞周期调控,衰老死亡等全部的生命活动。通过比较不同细胞类型的基因表达情况可以分析控制人类生命活动的生物学过程。当前对mRNA及其表达进行对比研究的方法已不止一种。包括消减文库筛选、差异杂交、代表性差异分析、基因表达序列分析、电子消减技术和DDRT-PCR技术等(以下简称DDRT-PCR)。目前以DDRT-PCR技术应用最为普遍[1]。 由于DDRT-PCR技术需要总RNA的量极少,操作简便、快速、灵敏,它在基因差异表达的研究方面的成功运用,使得DDRT-PCR被许多生物实验室作为一种重要工具,应用于体内和体外差异表达基因的筛选,研究基因功能[2]。 1DDRT-PCR技术的主要原理 DDRT-PCR技术以分子生物学上最广泛应用的两种技术PCR和聚丙烯酰胺凝胶电泳为基础。其基本原理是,以一对细胞(或组织)的总RNA反转录而成的cDNA为模板,利用PCR 的高效扩增,通过5′端与3′端引物的合理设计和组合,将细胞(或组织)中表达的约15000种基因片段直接显示在DNA测序胶上,从而找出一对细胞(或组织)中表达有差异的cDNA片段[2]。 由于人类基因组中只有3%的基因真正表达,因此通过研究mRNA的差异来研究基因的
实时荧光定量PCR(qPCR)技术简介
实时荧光定量 PCR 技术简介 实时荧光定量PCR(Quantitative Real-time PCR)是一项以PCR 反应为基础的DNA 定量技术,通过对目标基因在扩增过程中产生的拷贝数进行实时的定量,从而达到对目的基因 的定性和定量分析。现有两种常用的方法对PCR 产物进行荧光定量:一种是利用荧光染料 与双链DNA 结合,通过荧光强度进行定量;另一种是利用携带了荧光报告基团的特异DNA 探针对目标基因进行定量。 一、利用荧光染料进行定量 一种最为常用的定量方法就是在PCR 反应体系中加入荧光染料,此类荧光染料会与所 有的双链DNA 结合,并产生荧光。游离的荧光分子不会产生荧光信号,只有与双链DNA 结合的荧光分子才会释放荧光,随着DNA 拷贝数的增加,测得的荧光强度也会增强。 利用荧光染料进行定量的优势就是成本低廉,只需要一对普通的引物就能完成定量。然而,常用的诸如SYBR Green 染料会与所有的双链DNA 无差别地结合,包括引物二聚体,因 此有可能会导致对目标基因的定量不精确,灵敏度偏低。 二、利用荧光探针进行定量 荧光探针只能检测出与自身序列互补的DNA 片段,因此用探针法定量可以有效地避免引物二聚体的干扰,使定量结果更加精确。此外,通过使用携带不同荧光信号的多种探针,我 们可以同时对一个样品中的多个靶序列进行定量。 荧光探针的5’端携带有一个荧光报告基团,3’端则为淬灭基团,在正常情况下两个 基团间的距离很近,淬灭基团会抑制报告基团使其无法发出荧光。在PCR 反应过程中,引 物和荧光探针在退火阶段都会与目的片段结合;在延伸阶段,Taq 酶因为具有5’-3’核酸 外切酶活性,会将探针,使得报告基团和淬灭基团相互分开,从而释放出荧光。每增加一条目 的基因的拷贝,就会有一个探针被切开并释放荧光信号,因此随着PCR 反应的进行,荧光 信号会逐渐增强。 1 / 2
rtpcr原理
RT-PCR是将RNA的反转录(RT)和cDNA的聚合酶链式扩增(PCR)相结合的技术。 首先经反转录酶的作用从RNA合成 cDNA,再以cDNA为模板,扩增合成目的片段。RT-PCR技术灵敏而且用途广泛,可用于检测细胞中基因表达水平,细胞中RNA病毒的含量和直接克隆特定基因的cDNA序列。作为模板的RNA可以是总RNA、mRNA或体外转录的RNA产物。。RT-PCR用于对表达信息进行检测或定量。另外,这项技术还可以用来检测基因表达差异或不必构建cDNA文库克隆cDNA。RT-PCR比其他包括Northern印迹、RNase保护分析、原位杂交及S1核酸酶分析在内的RNA分析技术,更灵敏,更易于操作。逆转录反应可以使用逆转录酶,以随机引物、oligo(dT)或基因特异性的引物(GSP)起始。RT-PCR可以一步法或两步法的形式进行。在两步法RT-PCR中,每一步都在最佳条件下进行。cDNA的合成首先在逆转录缓冲液中进行,然后取出1/10的反应产物进行PCR。在一步法RT-PCR中,逆转录和PCR在同时为逆转录和PCR优化的条件下,在一只管中顺次进行。 逆转录酶(reverse transcriptase)是存在于RNA病毒体内的依赖RNA的DNA聚合酶,至少具有以下三种活性: 1、依赖RNA的DNA聚合酶活性:以RNA为模板合成cDNA第一条链 2、 Rnase水解活性:水解RNA杂合体中的RNA; 3、依赖DNA的DNA聚合酶活性:以第一条DNA链为模板合成互补的双链cDNA. 用于反转录的引物可视实验的具体情况选择随机引物、Oligo dT 及基因特异性引物中的一种。对于短的不具有发卡结构的真核细胞mRNA,三种都可。RT-PCR实验有三步:抽提RNA,RT,PCR。 试验前注意: 1. 做RT前必需测RNA浓度,逆转录体系对RNA量还是有一些要求,常用500ng或1ug 2. RT按要求做,一般不会出太大问题。 3. PCR如需扩长片段,则对前两步要求较高,需要有完整的cDNA存在,不是单改变Mg2+浓度、退火温度能解决的。 引物合成
四大大屏显示技术对比分析
四大大屏显示技术对比分析 随着信息化技术的提高,人们对于视觉欣赏的要求越来越高。“视觉冲击力”成为人们评判显示性能的一个标准。视觉冲击力不仅来自于清晰地画面,还来自于超大尺寸的画面。为了满足这种诉求,大屏拼接应运而生。此外,能实现超大画面的还有基于投影技术的边缘融合技术。从目前的情况来看,在超大图像的应用方面,各项技术是“各花入各眼”,在不同的领域发挥着各种不同的优势。 大屏拼接系统 目前,比较常见的大屏幕拼接系统,通常根据显示单元的工作方式分为三个主要类型,即LCD显示单元拼接、PDP显示单元拼接和DLP背投显示单元拼接。其中前二者属于平板显示单元拼接系统,后者属于投影单元拼接系统。 等离子大屏拼接系统 PDP ( Plasma Display Panel ),即等离子显示屏。PDP是一种利用气体放电的显示技术,其工作原理与日光灯很相似。它采用了等离子管作为发光元件,屏幕上每一个等离子管对应一个像素,屏幕以玻璃作为基板,基板间隔一定距离,形成一个个放电空间。放电空间内充入氖、氙等混合惰性气体作为工作媒质,在两块玻璃基板的内侧面上涂有金属氧化物导电薄膜作激励电极。当向电极上加入电压,放电空间内的混合气体便发生等离子体放电现象,也称电浆效应。等离子体放电产生紫外线,紫外线激发涂有红绿蓝荧光粉的荧光屏,荧光屏发射出可见光,显现出图像。 PDP单元拼接具有颜色鲜亮、高对比度以及高亮度的优点,同时也具有其自身无法克服的缺点。等离子由于耗电量与发热量很大,会产生严重灼伤现象,并不适用于长期静态画面显示监控。并且PDP单元用于拼接之后,整机升温更高,致使设备容易烧毁。此外,目前市面上等离子拼接幕墙价格较高,一般一平方米的价格高达十几万。今天,在低碳、节能已经成为主流趋势,对于大多数普通用户来说,等离子拼接显然不是其最优选择。 尽管缺点多多,然而等离子颜色鲜亮、高亮度的特性使得其画面显示效果具有突出的优势,这使得等离子拼接成为一些展览展示活动的宠儿。另外,对于画面质量要求较高的政府机关等离子也是首选。不过,从整体市场的占有率来说,等离子拼接处于完全的劣势,而且就整个行业的发展趋势来说,等离子拼接的发展潜力有限。据部分业内人士分析,该行业目前是“鸡肋产业”,未来肯定会被取代。 LCD液晶拼接 所谓的LCD液晶大屏拼接,是采用LCD显示单元拼接的方式,通过拼接控制软件系统,来实现大屏幕显示效果的一种拼接屏体。LCD液晶拼接目前以韩国三星的DID为代表,虽然拼接市场上还有SHARP、LG、NEC等品牌,但是DID以其优良的性价比在LCD平板拼接技术中一枝独秀。 LCD拼接具有厚度薄、重量轻、低能耗、长寿命、无辐射等优点,而且其画面细腻、分辨率高,各项关键性能指标的优秀表现,已使它成为发展主流,前景看好。作为拼接显示终端,LCD尽管有上述优点,但是作为拼接显示单元,其缺点也是致命的:那就是目前其拼缝教大,且在三种拼接显示单元中最大,令许多用户不得不忍痛舍弃。 就目前的发展态势来看,作为新生技术,尽管LCD拼接存在严重的缺陷,但是大屏拼接业内大多数人士还是对其寄予厚望。如今大屏拼接系统的发展需求已经日益明朗——那就
OLED显示技术和LCD液晶显示技术的对比介绍资料
OLED显示技术和LCD液晶显示技术的对比介绍今年开年的新品,个家都开始做曲面电视,在这之前很多厂家就打出了OLED 电视的旗号,那么这OLED就是曲面电视?和液晶电视有啥具体的区别? 一般我们将的液晶电视,一般讲的就是LCD电视,LCD 电视是 Liquid Crystal Display 的简称,是液晶显示屏的全称:它包括了TFT,UFB,TFD,STN 等类型的液晶显示屏。 LCD 的构造是在两片平行的玻璃当中放置液态的晶体,两片玻璃中间有许多垂直和水平的细小电线,透过通电与否来控制杆状水晶分子改变方向,将光线折射出来产生画面。 而OLED电视,已经不再需要LCD液晶面板,RGB色彩信号直接由OLED二极管显示,几乎已经不存在液晶的可视角度问题,2013年1月,LG率先推出了采用OLED的55寸电视,但价格昂贵,短时间内还无法大规模普及。 2016年可以说是OLED代表年,其投入能量之多、士气之疯狂,宣传之声势浩大,仿佛就要攻破LCD坚守十年的围墙!现在中日台韩面板厂一字排开,每个都摇着OLED的旗子,旗海飘扬绵延到天边,仿佛宣告着显示技术的世代交替山雨欲来,史诺也曾经一度迷惘,难道“后液晶时代”提早来了吗? 放眼过去数年,OLED只是雄踞一方,虽然排挤了一点LCD的疆界,但几年下来也相安无事。OLED势力有两大部落,分别叫“三星”跟“LG”,一直以来他们约定好,三星你玩小尺寸、LG我玩大尺寸,三星你玩RGB OLED,LG我来弄弄White OLED,偶而可以跨对方的界,但浅尝则止,说好不能太认真。 还有就是OLED做出来后,咱们不外卖也不外购,坚持“自己的OLED自己做”,于是金星联盟就这样自成一格、好不快乐,自产自销用整机出口帮自己练功,LCD也只能眼巴巴的望着,想着:“还好嘛!不过就是自发光阿,说来萤火虫也是自发光嘛,我还怕虫子咬不成?”。 时光快转到2015年下半,金星连线开始外销,还在中国成立了OLED联盟,中国彩电跟智慧手机品牌一个接一个开出OLED旗舰,LCD才在心里想着不妙,好像该做点什么,结果转身一看,苹果公司库克也宣布将要采用OLED的显示屏幕。 “完了,一切都完了。”LCD一瞬间觉得自己成了反派。好的,说到这里